

Spleen Tyrosine Kinase phosphorylates VE-cadherin to cause endothelial barrier disruption in acute lung injury
- PMID: 38229397
- PMCID: PMC10731244
- DOI: 10.1016/j.jbc.2023.105408
Spleen Tyrosine Kinase phosphorylates VE-cadherin to cause endothelial barrier disruption in acute lung injury
Abstract
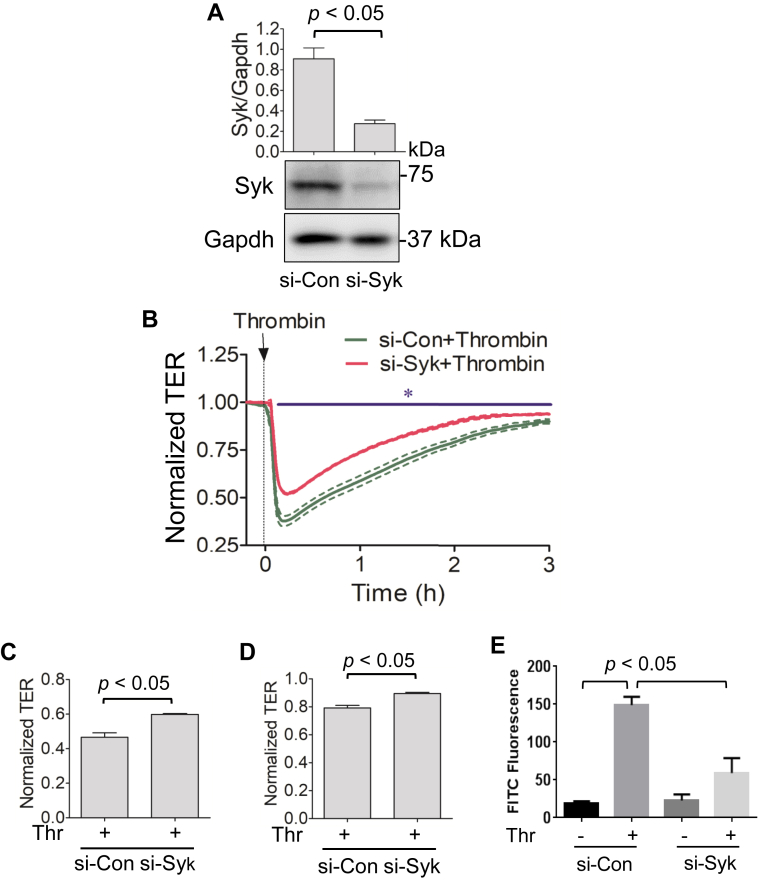
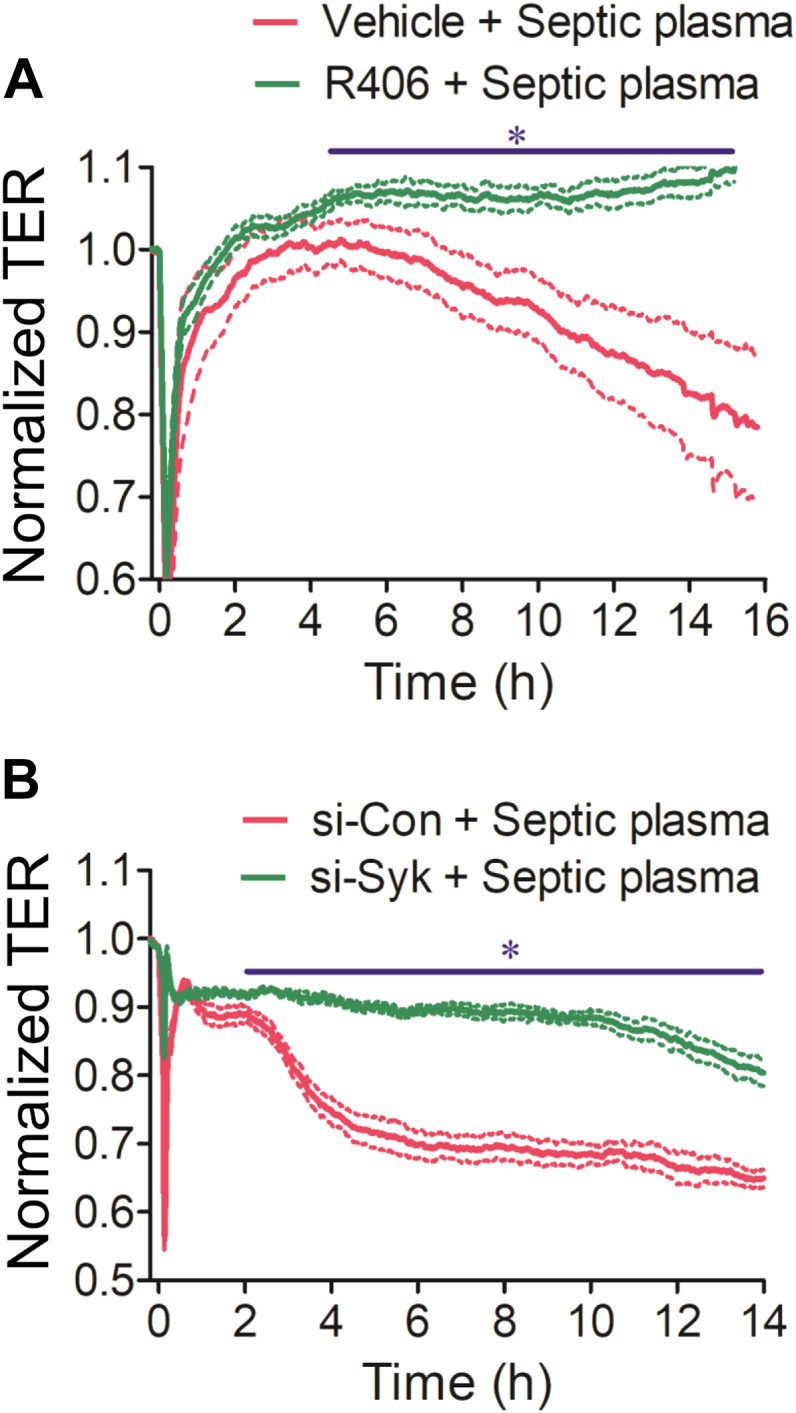
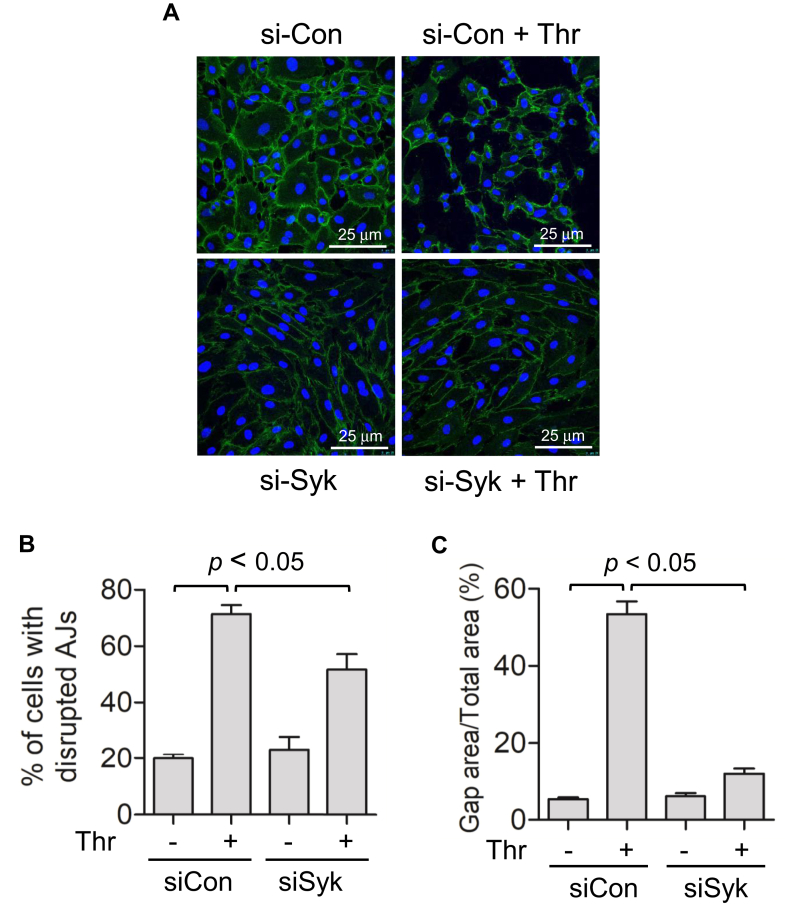
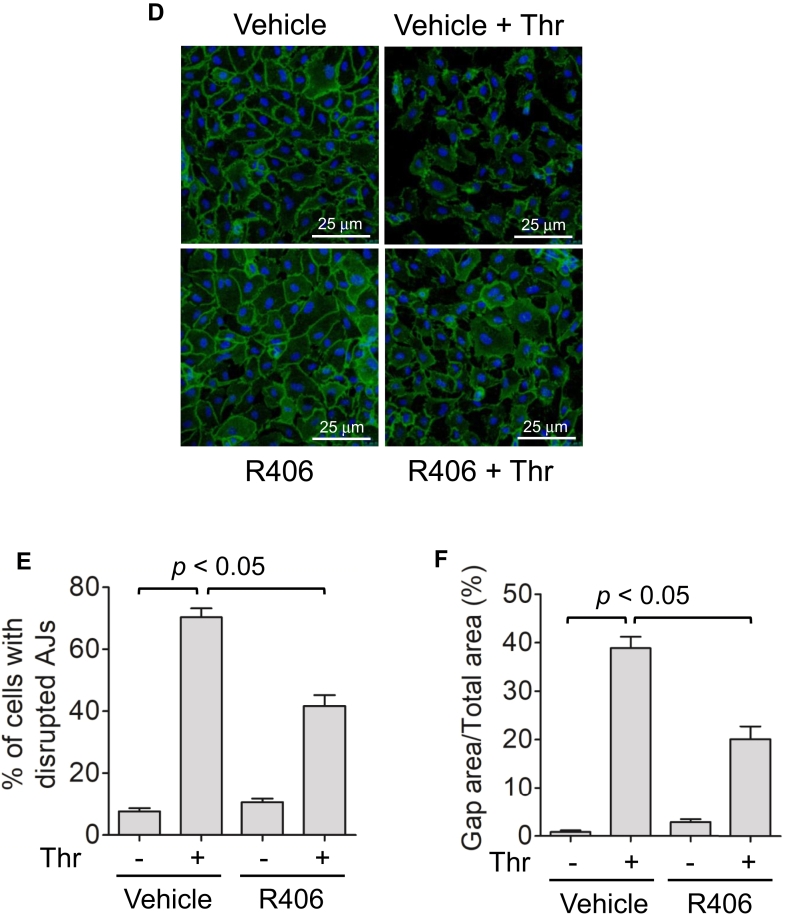
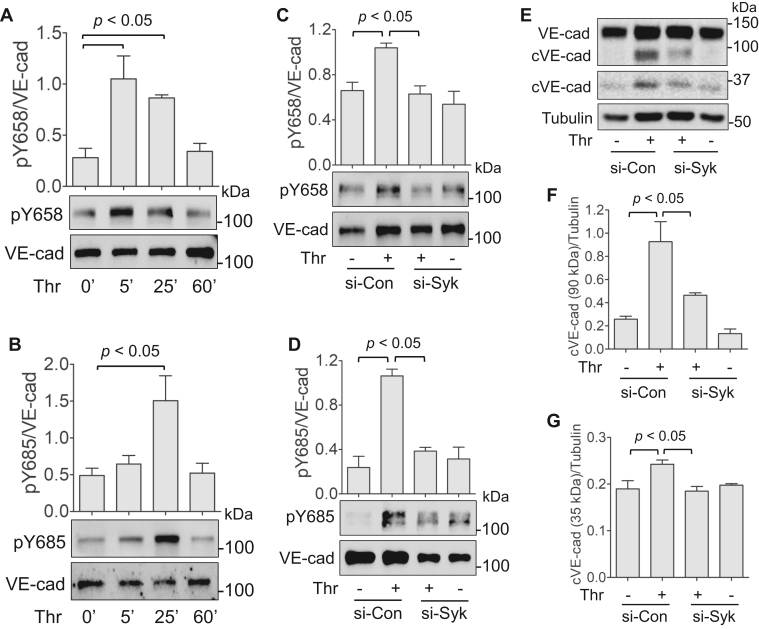
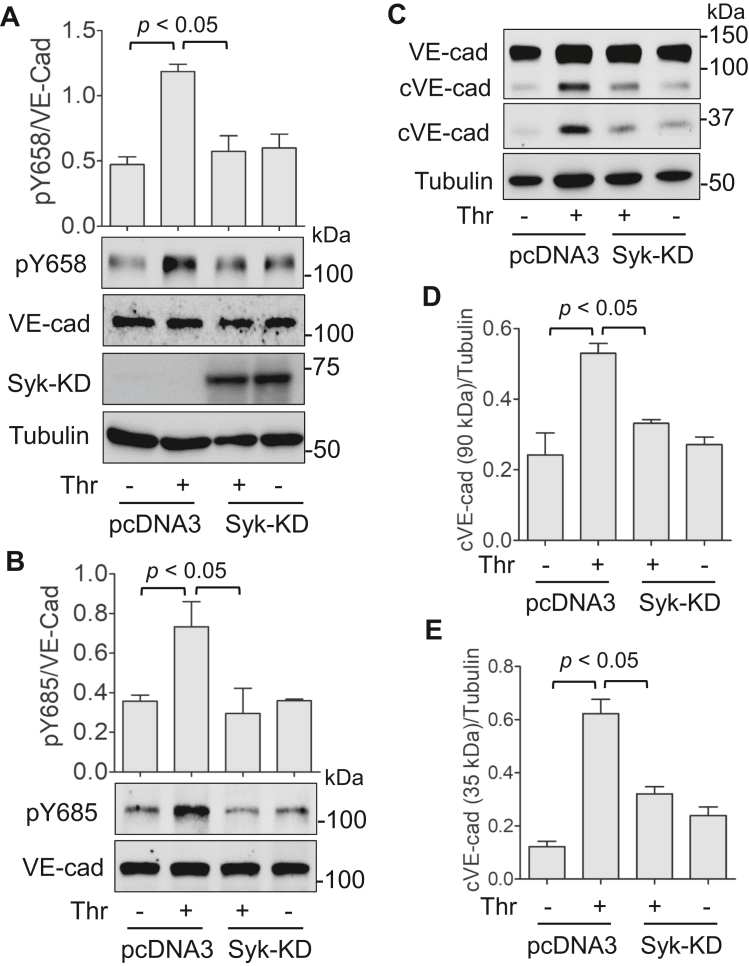
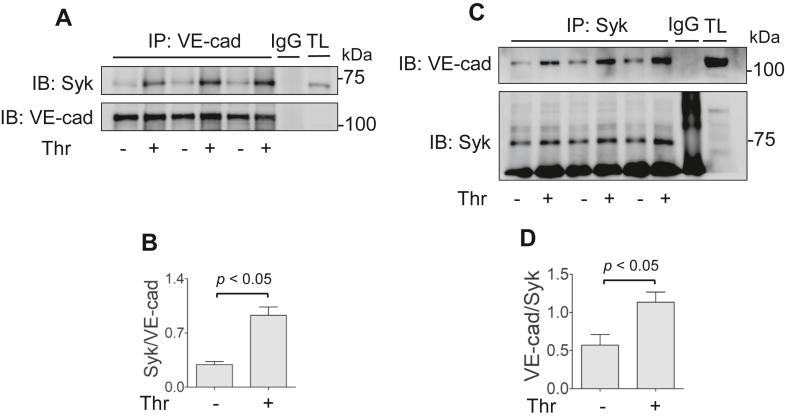
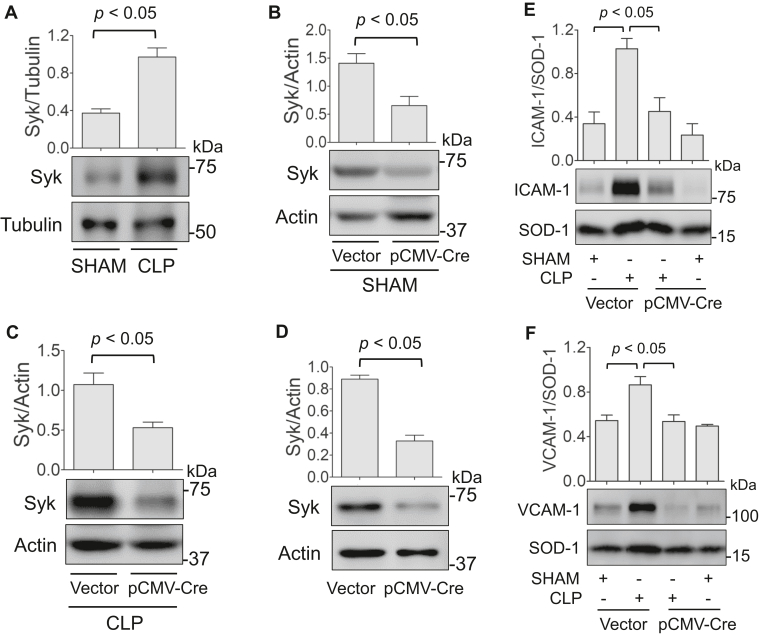
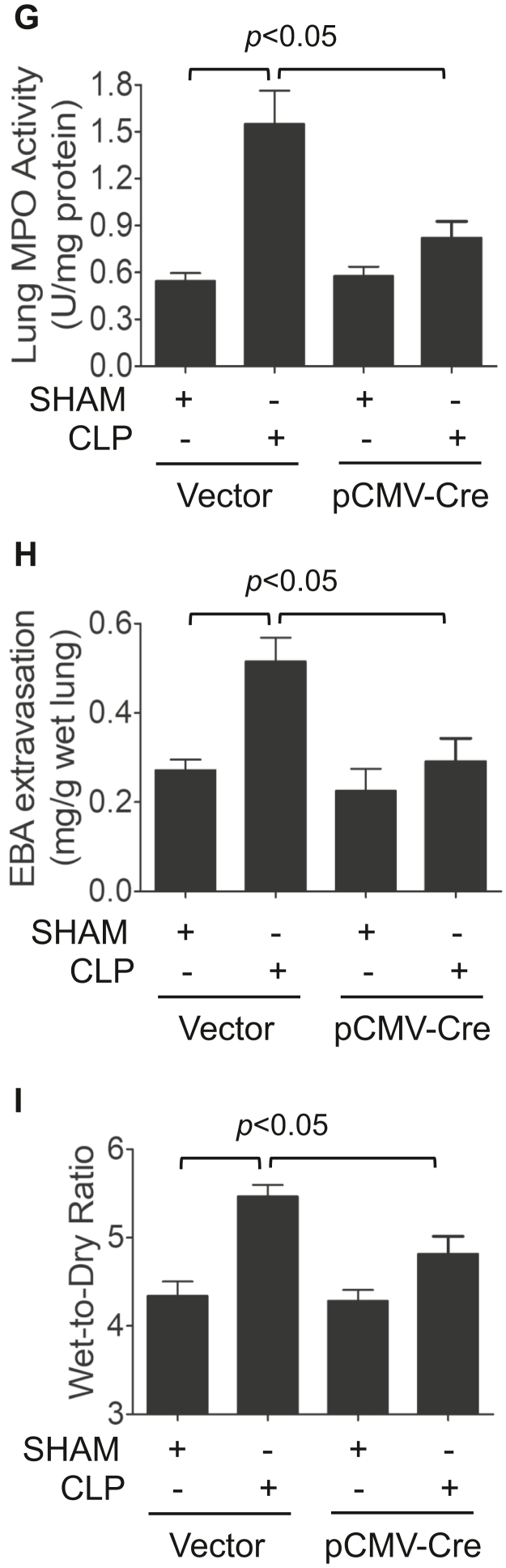
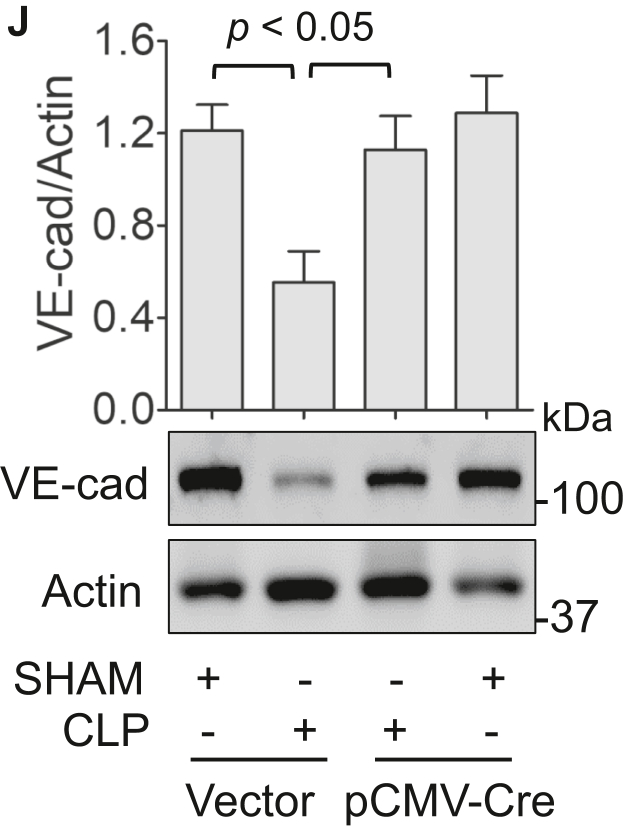
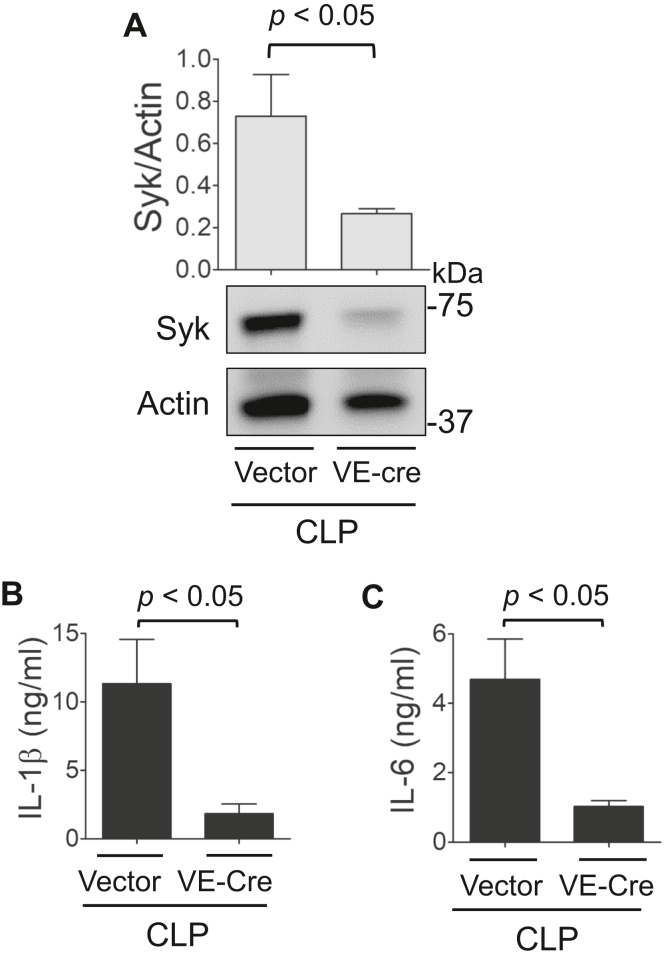
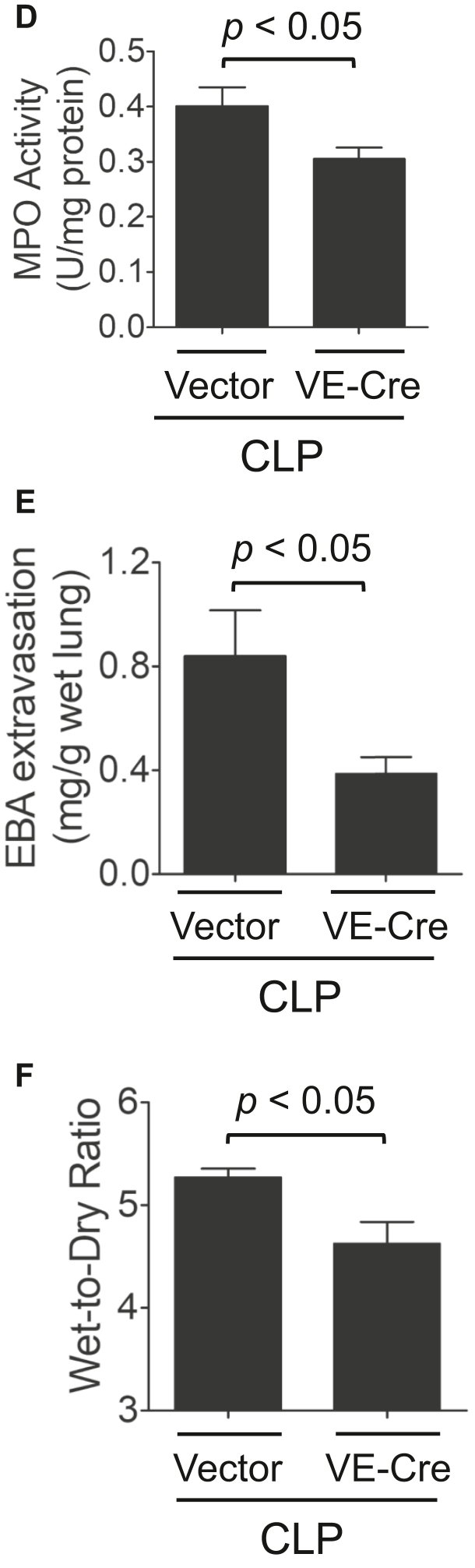
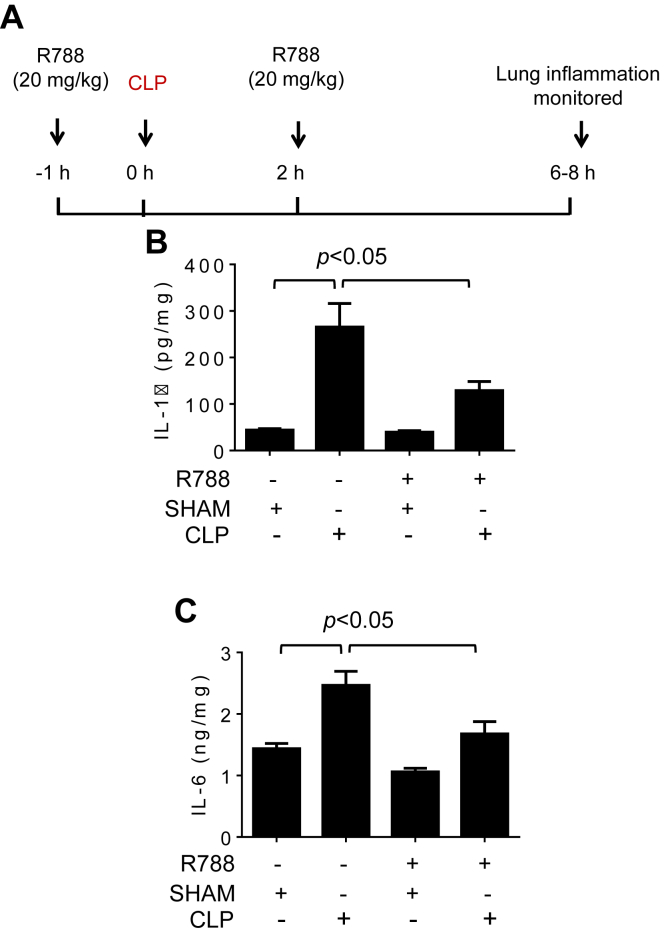
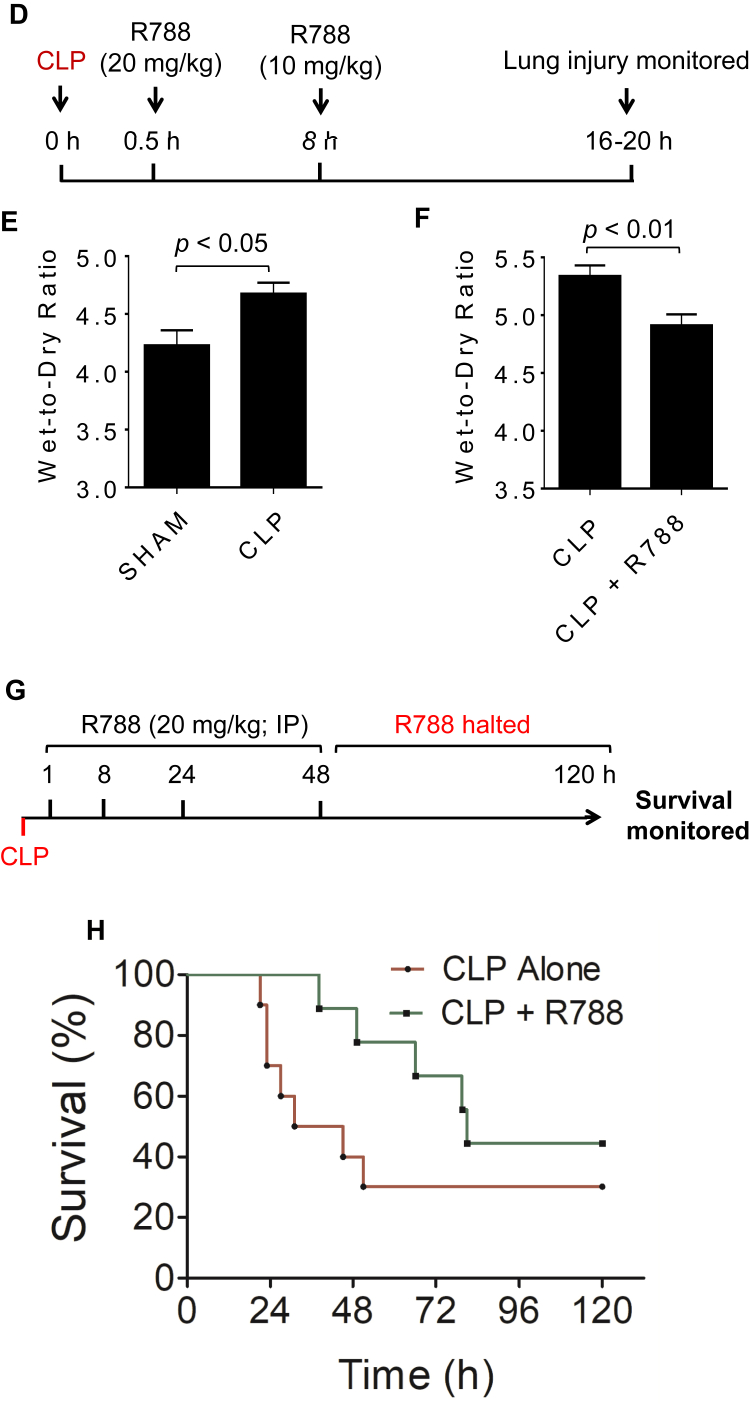
Increased endothelial cell (EC) permeability is a cardinal feature of acute lung injury/acute respiratory distress syndrome (ALI/ARDS). Tyrosine phosphorylation of VE-cadherin is a key determinant of EC barrier disruption. However, the identity and role of tyrosine kinases in this context are incompletely understood. Here we report that Spleen Tyrosine Kinase (Syk) is a key mediator of EC barrier disruption and lung vascular leak in sepsis. Inhibition of Syk by pharmacological or genetic approaches, each reduced thrombin-induced EC permeability. Mechanistically, Syk associates with and phosphorylates VE-cadherin to cause EC permeability. To study the causal role of endothelial Syk in sepsis-induced ALI, we used a remarkably efficient and cost-effective approach based on gene transfer to generate EC-ablated Syk mice. These mice were protected against sepsis-induced loss of VE-cadherin and inflammatory lung injury. Notably, the administration of Syk inhibitor R788 (fostamatinib); currently in phase II clinical trial for the treatment of COVID-19, mitigated lung injury and mortality in mice with sepsis. These data identify Syk as a novel kinase for VE-cadherin and a druggable target against ALI in sepsis.
Keywords: Syk; VE-cadherin; endothelial cells; lung vascular leak; sepsis.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
MTOR maintains endothelial cell integrity to limit lung vascular injury.J Biol Chem. 2024 Dec;300(12):107952. doi: 10.1016/j.jbc.2024.107952. Epub 2024 Nov 6. J Biol Chem. 2024. PMID: 39510184 Free PMC article.
-
Select Rab GTPases Regulate the Pulmonary Endothelium via Endosomal Trafficking of Vascular Endothelial-Cadherin.Am J Respir Cell Mol Biol. 2016 Jun;54(6):769-81. doi: 10.1165/rcmb.2015-0286OC. Am J Respir Cell Mol Biol. 2016. PMID: 26551054 Free PMC article.
-
Bone morphogenetic protein 10 serves as a biomarker and a potential therapeutic target for endothelial dysfunction in endotoxin-induced acute lung injury.J Transl Med. 2025 Jul 8;23(1):755. doi: 10.1186/s12967-025-06742-6. J Transl Med. 2025. PMID: 40629429 Free PMC article.
-
Pressure-controlled versus volume-controlled ventilation for acute respiratory failure due to acute lung injury (ALI) or acute respiratory distress syndrome (ARDS).Cochrane Database Syst Rev. 2015 Jan 14;1(1):CD008807. doi: 10.1002/14651858.CD008807.pub2. Cochrane Database Syst Rev. 2015. PMID: 25586462 Free PMC article.
-
Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications.Cell Commun Signal. 2024 Apr 25;22(1):241. doi: 10.1186/s12964-024-01620-y. Cell Commun Signal. 2024. PMID: 38664775 Free PMC article. Review.
Cited by
-
Vascular syk-ness: A new role for an old immunological favorite.J Biol Chem. 2024 Jul;300(7):107517. doi: 10.1016/j.jbc.2024.107517. Epub 2024 Jun 28. J Biol Chem. 2024. PMID: 38945448 Free PMC article.
-
Spleen tyrosine kinase: a novel pharmacological target for sepsis-induced cardiac dysfunction and multi-organ failure.Front Immunol. 2024 Nov 4;15:1447901. doi: 10.3389/fimmu.2024.1447901. eCollection 2024. Front Immunol. 2024. PMID: 39559354 Free PMC article.
-
MTOR maintains endothelial cell integrity to limit lung vascular injury.J Biol Chem. 2024 Dec;300(12):107952. doi: 10.1016/j.jbc.2024.107952. Epub 2024 Nov 6. J Biol Chem. 2024. PMID: 39510184 Free PMC article.
References
-
- Levitt J.E., Matthay M.A. Treatment of acute lung injury: historical perspective and potential future therapies. Semin. Respir. Crit. Care Med. 2006;27:426–437. - PubMed
-
- Bhattacharya J., Matthay M.A. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu. Rev. Physiol. 2013;75:593–615. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
