

scMAE: a masked autoencoder for single-cell RNA-seq clustering
- PMID: 38230824
- PMCID: PMC10832357
- DOI: 10.1093/bioinformatics/btae020
scMAE: a masked autoencoder for single-cell RNA-seq clustering
Abstract
Motivation: Single-cell RNA sequencing has emerged as a powerful technology for studying gene expression at the individual cell level. Clustering individual cells into distinct subpopulations is fundamental in scRNA-seq data analysis, facilitating the identification of cell types and exploration of cellular heterogeneity. Despite the recent development of many deep learning-based single-cell clustering methods, few have effectively exploited the correlations among genes, resulting in suboptimal clustering outcomes.
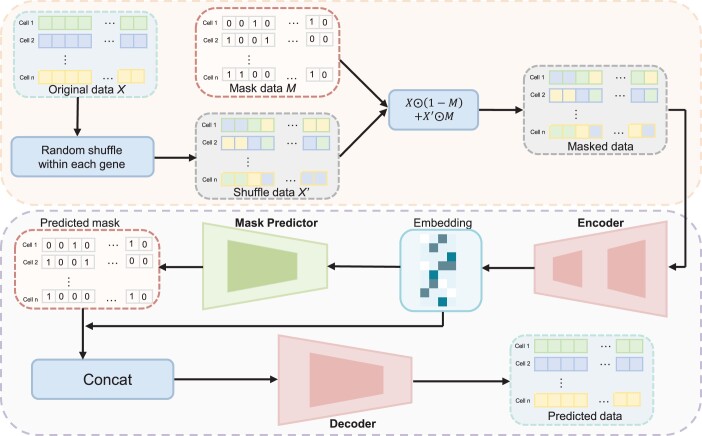
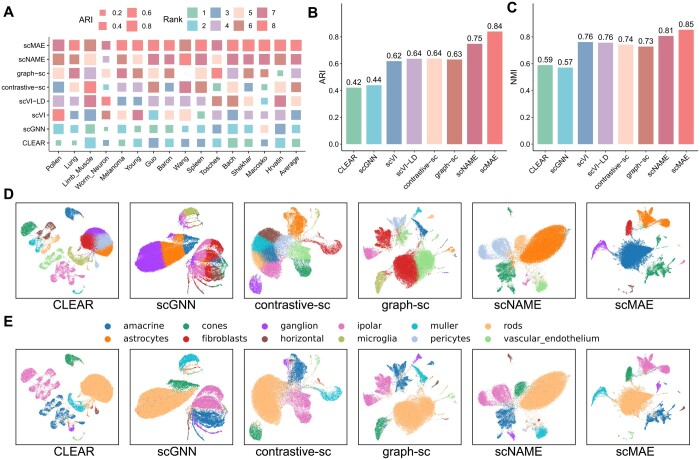
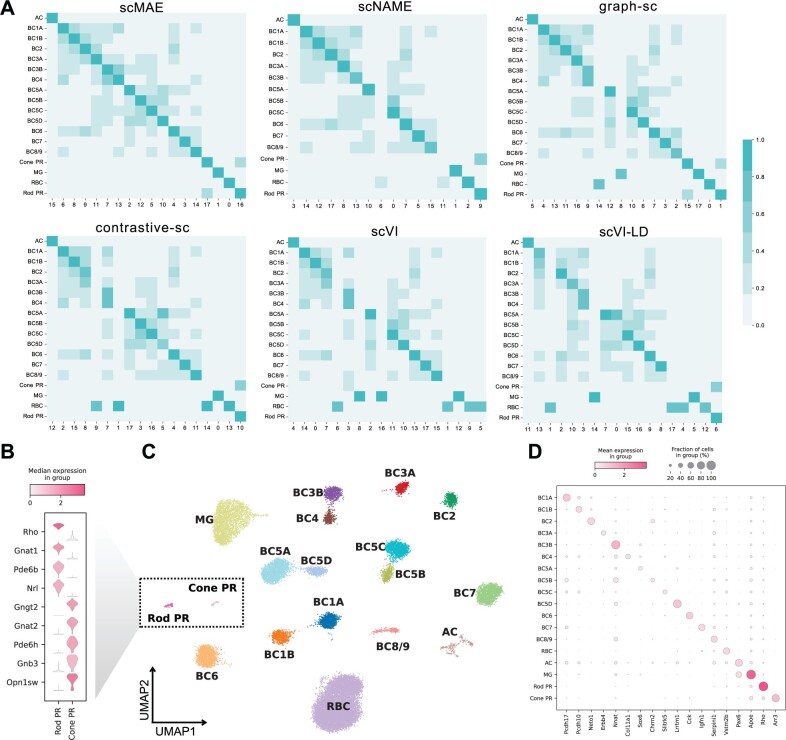
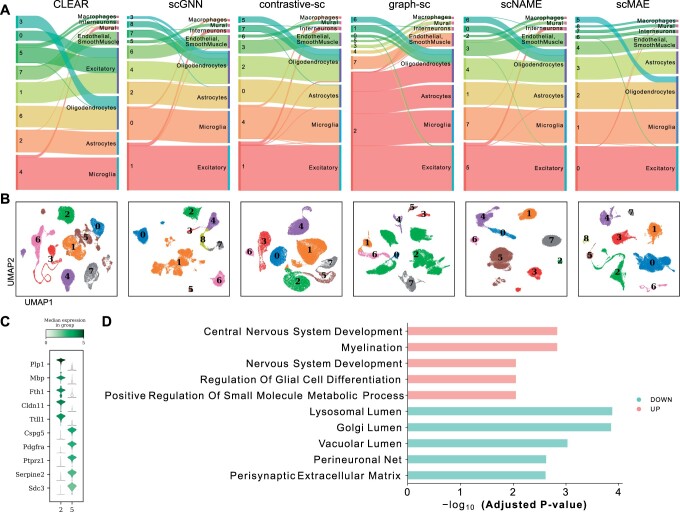
Results: Here, we propose a novel masked autoencoder-based method, scMAE, for cell clustering. scMAE perturbs gene expression and employs a masked autoencoder to reconstruct the original data, learning robust and informative cell representations. The masked autoencoder introduces a masking predictor, which captures relationships among genes by predicting whether gene expression values are masked. By integrating this masking mechanism, scMAE effectively captures latent structures and dependencies in the data, enhancing clustering performance. We conducted extensive comparative experiments using various clustering evaluation metrics on 15 scRNA-seq datasets from different sequencing platforms. Experimental results indicate that scMAE outperforms other state-of-the-art methods on these datasets. In addition, scMAE accurately identifies rare cell types, which are challenging to detect due to their low abundance. Furthermore, biological analyses confirm the biological significance of the identified cell subpopulations.
Availability and implementation: The source code of scMAE is available at: https://zenodo.org/records/10465991.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Blondel VD, Guillaume J-L, Lambiotte R. et al. Fast unfolding of communities in large networks. J Stat Mech 2008;2008:P10008.
-
- Buettner F, Natarajan KN, Casale FP. et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol 2015;33:155–60. - PubMed
