

This is a preprint.
Signatures of transmission in within-host M. tuberculosis variation
- PMID: 38234741
- PMCID: PMC10793532
- DOI: 10.1101/2023.12.28.23300451
Signatures of transmission in within-host M. tuberculosis variation
Update in
-
Signatures of transmission in within-host Mycobacterium tuberculosis complex variation: a retrospective genomic epidemiology study.Lancet Microbe. 2025 Jan;6(1):100936. doi: 10.1016/j.lanmic.2024.06.003. Epub 2024 Nov 28. Lancet Microbe. 2025. PMID: 39615498 Free PMC article.
Abstract
Background: Because M. tuberculosis evolves slowly, transmission clusters often contain multiple individuals with identical consensus genomes, making it difficult to reconstruct transmission chains. Finding additional sources of shared M. tuberculosis variation could help overcome this problem. Previous studies have reported M. tuberculosis diversity within infected individuals; however, whether within-host variation improves transmission inferences remains unclear.
Methods: To evaluate the transmission information present in within-host M. tuberculosis variation, we re-analyzed publicly available sequence data from three household transmission studies, using household membership as a proxy for transmission linkage between donor-recipient pairs.
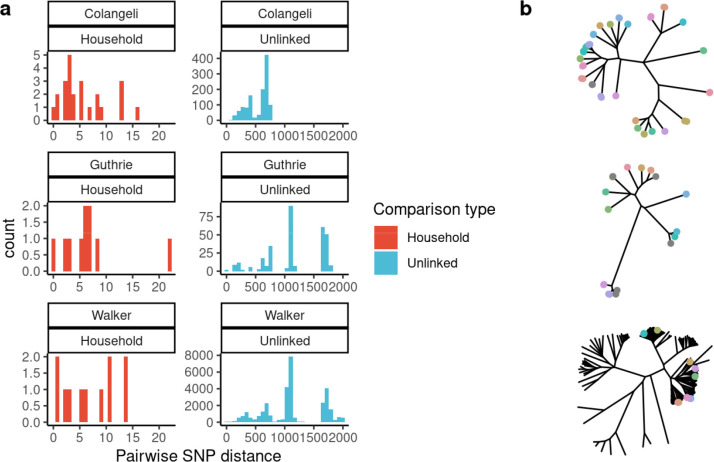
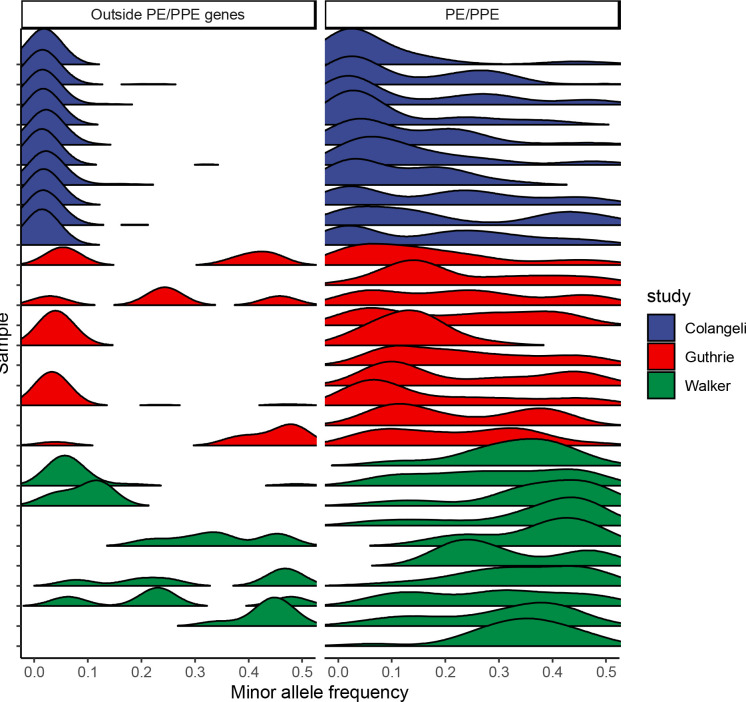
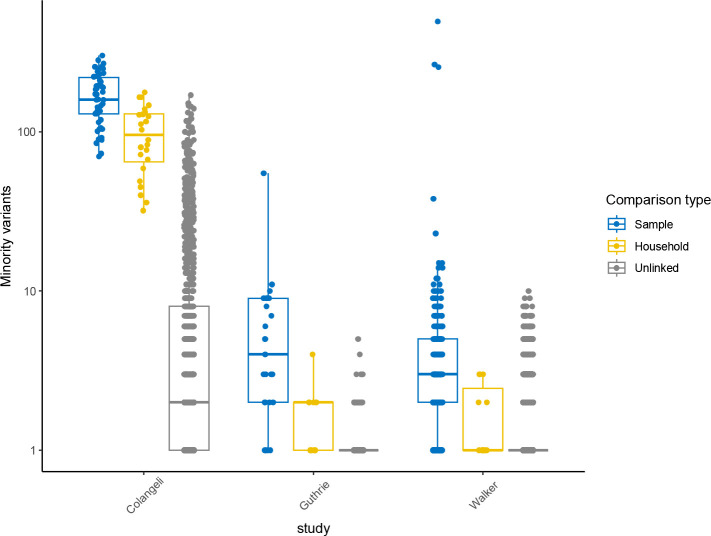
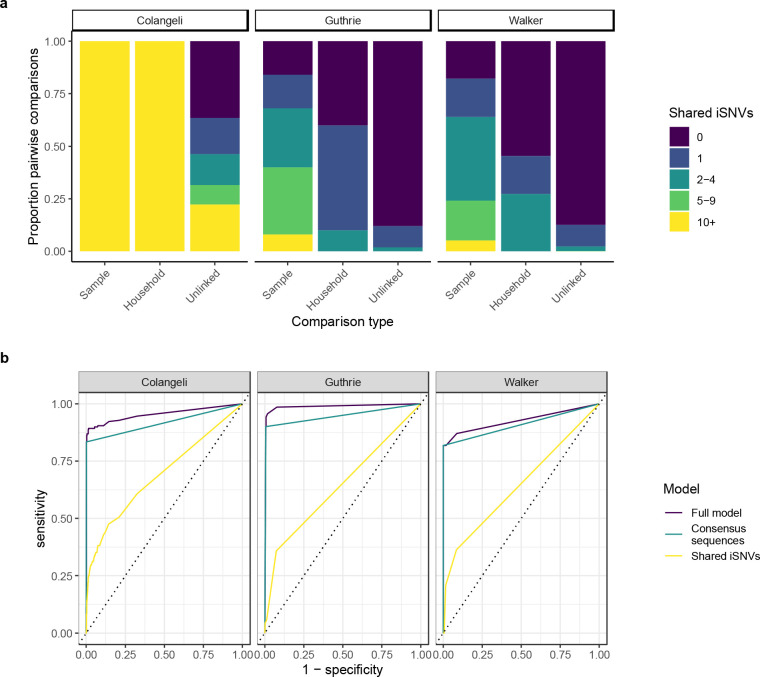
Findings: We found moderate levels of minority variation present in M. tuberculosis sequence data from cultured isolates that varied significantly across studies (mean: 6, 7, and 170 minority variants above a 1% minor allele frequency threshold, outside of PE/PPE genes). Isolates from household members shared more minority variants than did isolates from unlinked individuals in the three studies (mean 98 shared minority variants vs. 10; 0.8 vs. 0.2, and 0.7 vs. 0.2, respectively). Shared within-host variation was significantly associated with household membership (OR: 1.51 [1.30,1.71], for one standard deviation increase in shared minority variants). Models that included shared within-host variation improved the accuracy of predicting household membership in all three studies as compared to models without within-host variation (AUC: 0.95 versus 0.92, 0.99 versus 0.95, and 0.93 versus 0.91).
Interpretation: Within-host M. tuberculosis variation persists through culture and could enhance the resolution of transmission inferences. The substantial differences in minority variation recovered across studies highlights the need to optimize approaches to recover and incorporate within-host variation into automated phylogenetic and transmission inference.
Funding: NIAID: 5K01AI173385.
Keywords: M. tuberculosis; epidemiology; genomics; transmission.
Conflict of interest statement
Declaration of interest The authors report no conflict of interest.
Figures
References
-
- Yates T. A. et al. The transmission of Mycobacterium tuberculosis in high burden settings. Lancet Infect Dis 16, 227–238 (2016). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources