

Sotorasib Is a Pan-RASG12C Inhibitor Capable of Driving Clinical Response in NRASG12C Cancers
- PMID: 38236605
- PMCID: PMC11061598
- DOI: 10.1158/2159-8290.CD-23-1138
Sotorasib Is a Pan-RASG12C Inhibitor Capable of Driving Clinical Response in NRASG12C Cancers
Abstract
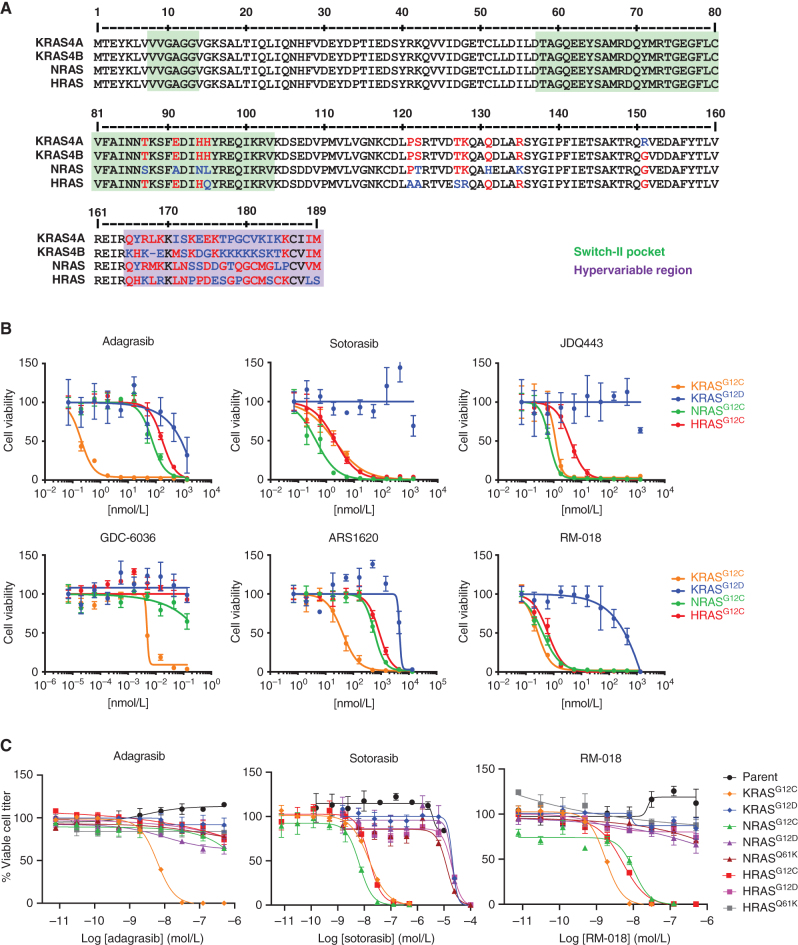
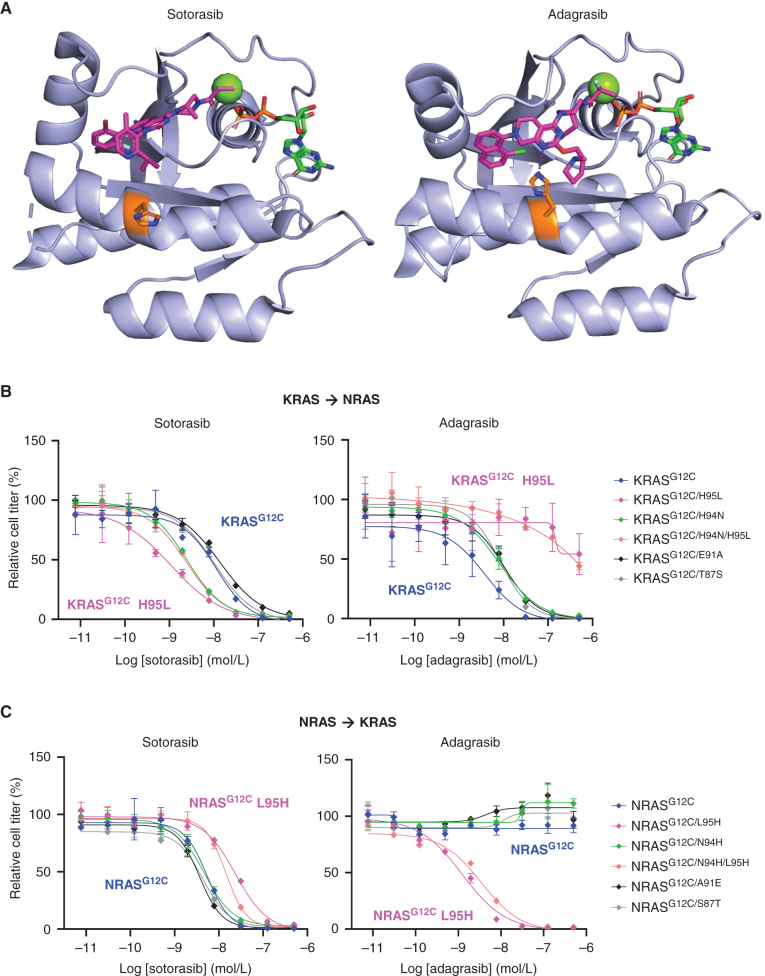
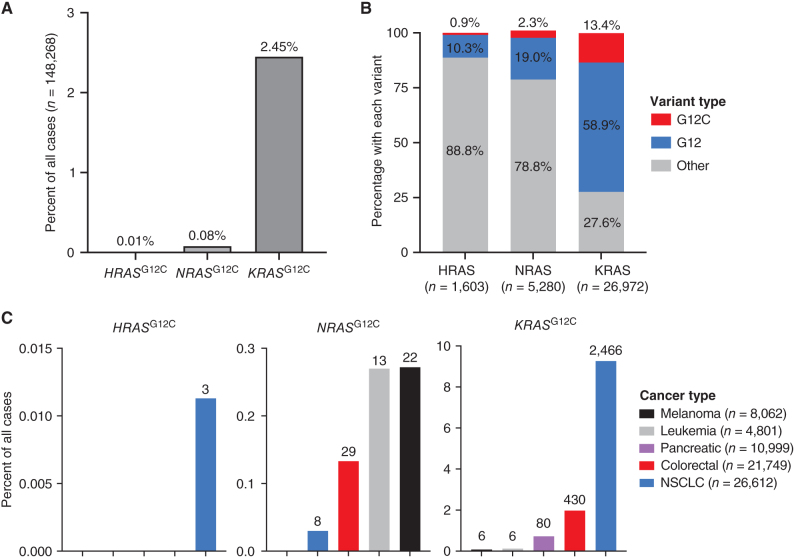
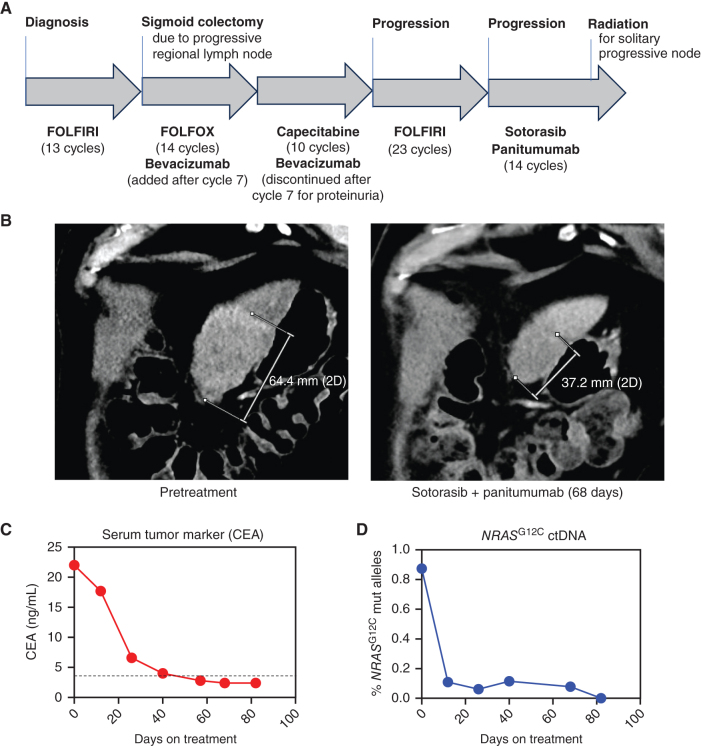
KRASG12C inhibitors, like sotorasib and adagrasib, potently and selectively inhibit KRASG12C through a covalent interaction with the mutant cysteine, driving clinical efficacy in KRASG12C tumors. Because amino acid sequences of the three main RAS isoforms-KRAS, NRAS, and HRAS-are highly similar, we hypothesized that some KRASG12C inhibitors might also target NRASG12C and/or HRASG12C, which are less common but critical oncogenic driver mutations in some tumors. Although some inhibitors, like adagrasib, were highly selective for KRASG12C, others also potently inhibited NRASG12C and/or HRASG12C. Notably, sotorasib was five-fold more potent against NRASG12C compared with KRASG12C or HRASG12C. Structural and reciprocal mutagenesis studies suggested that differences in isoform-specific binding are mediated by a single amino acid: Histidine-95 in KRAS (Leucine-95 in NRAS). A patient with NRASG12C colorectal cancer treated with sotorasib and the anti-EGFR antibody panitumumab achieved a marked tumor response, demonstrating that sotorasib can be clinically effective in NRASG12C-mutated tumors.
Significance: These studies demonstrate that certain KRASG12C inhibitors effectively target all RASG12C mutations and that sotorasib specifically is a potent NRASG12C inhibitor capable of driving clinical responses. These findings have important implications for the treatment of patients with NRASG12C or HRASG12C cancers and could guide design of NRAS or HRAS inhibitors. See related commentary by Seale and Misale, p. 698. This article is featured in Selected Articles from This Issue, p. 695.
©2024 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol 2018;15:709–20. - PubMed
-
- Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. . Adagrasib in Non-small-cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med 2022;387:120–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
