

Longitudinal single cell atlas identifies complex temporal relationship between type I interferon response and COVID-19 severity
- PMID: 38238298
- PMCID: PMC10796319
- DOI: 10.1038/s41467-023-44524-0
Longitudinal single cell atlas identifies complex temporal relationship between type I interferon response and COVID-19 severity
Abstract
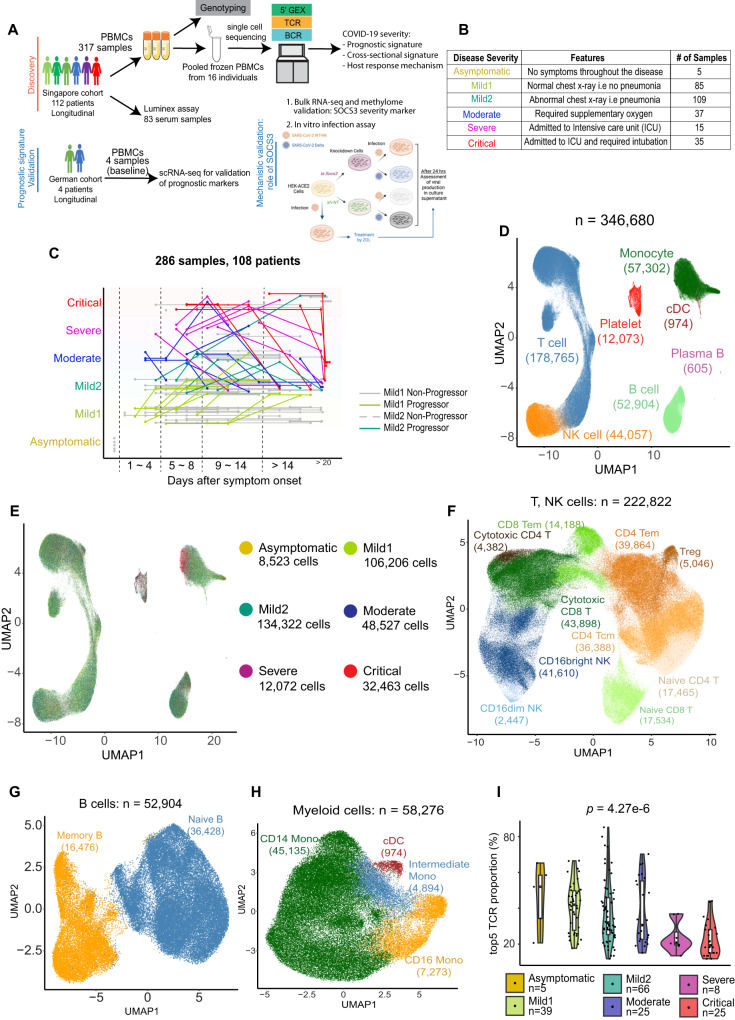
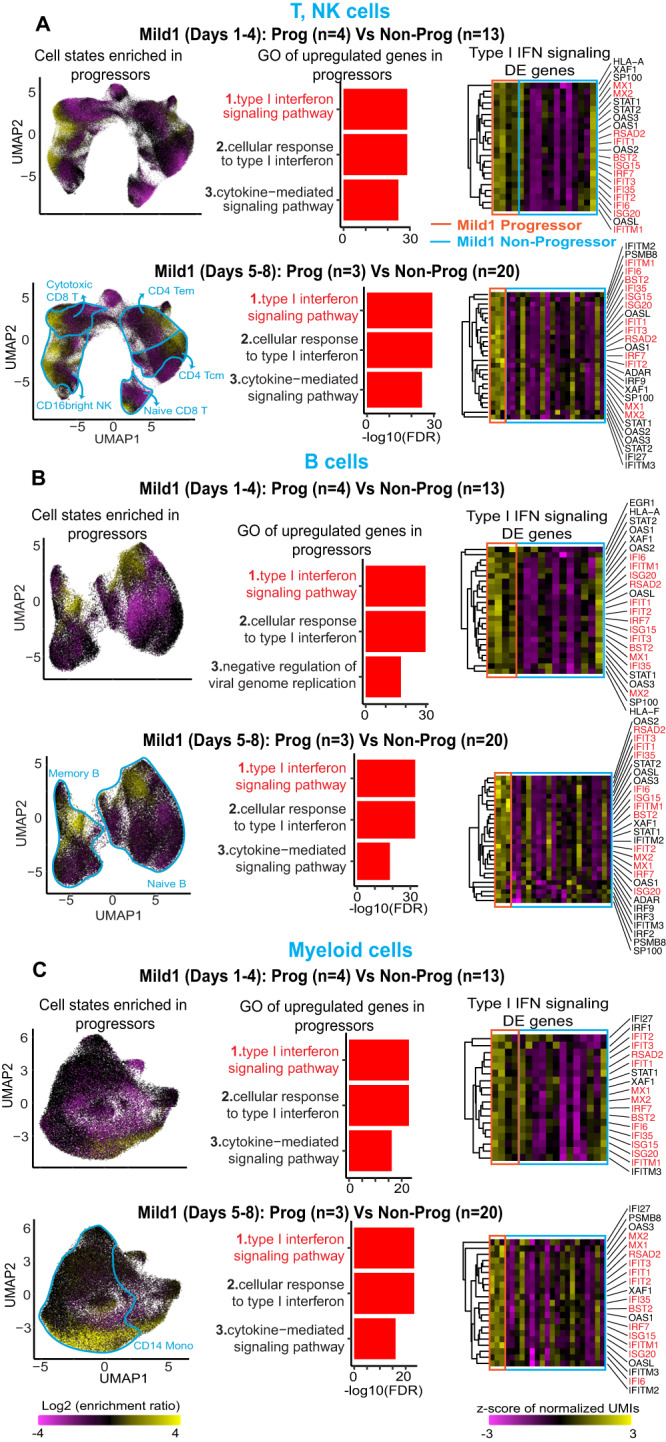
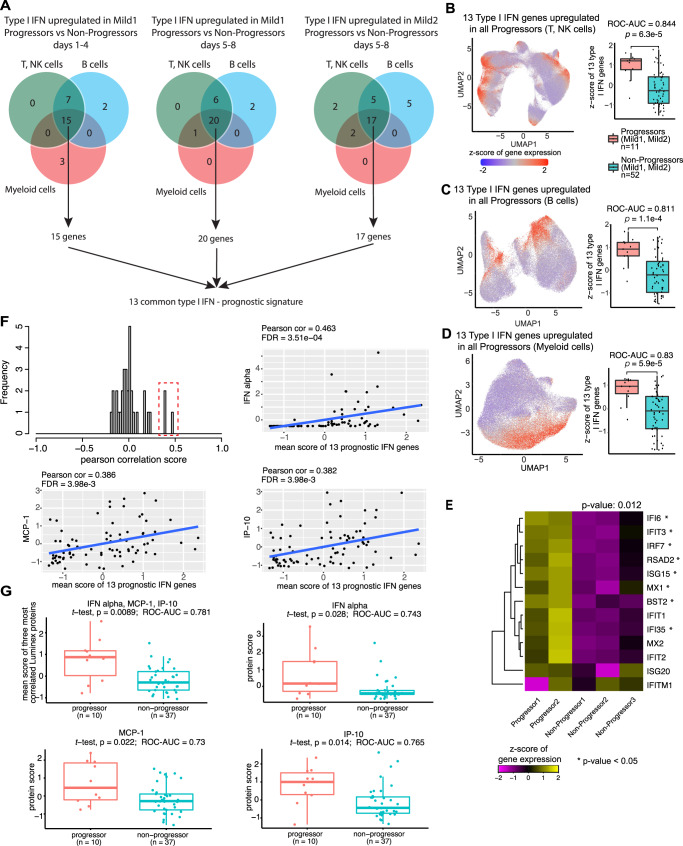
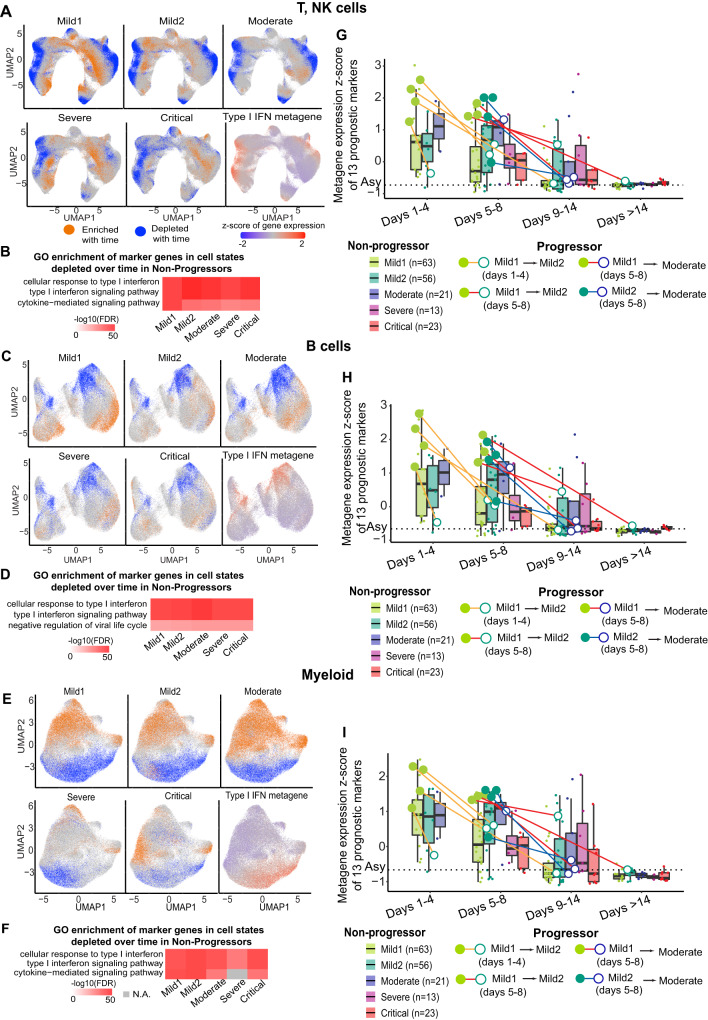
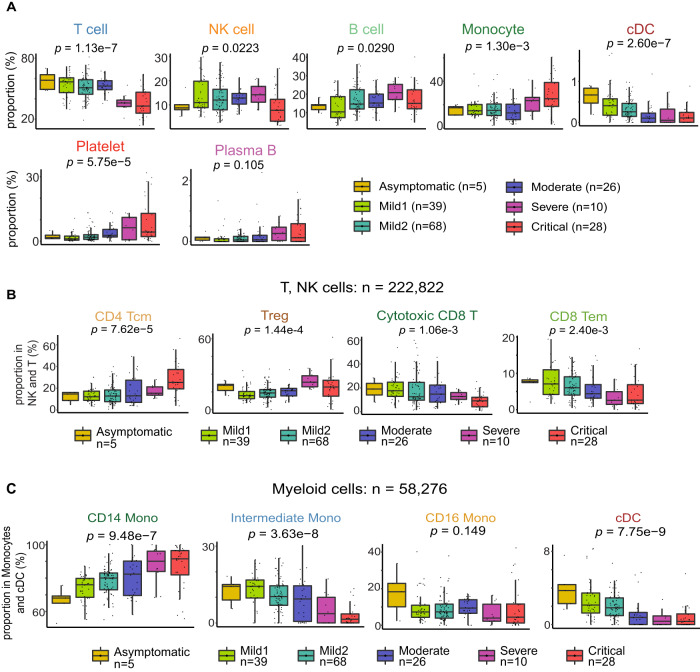
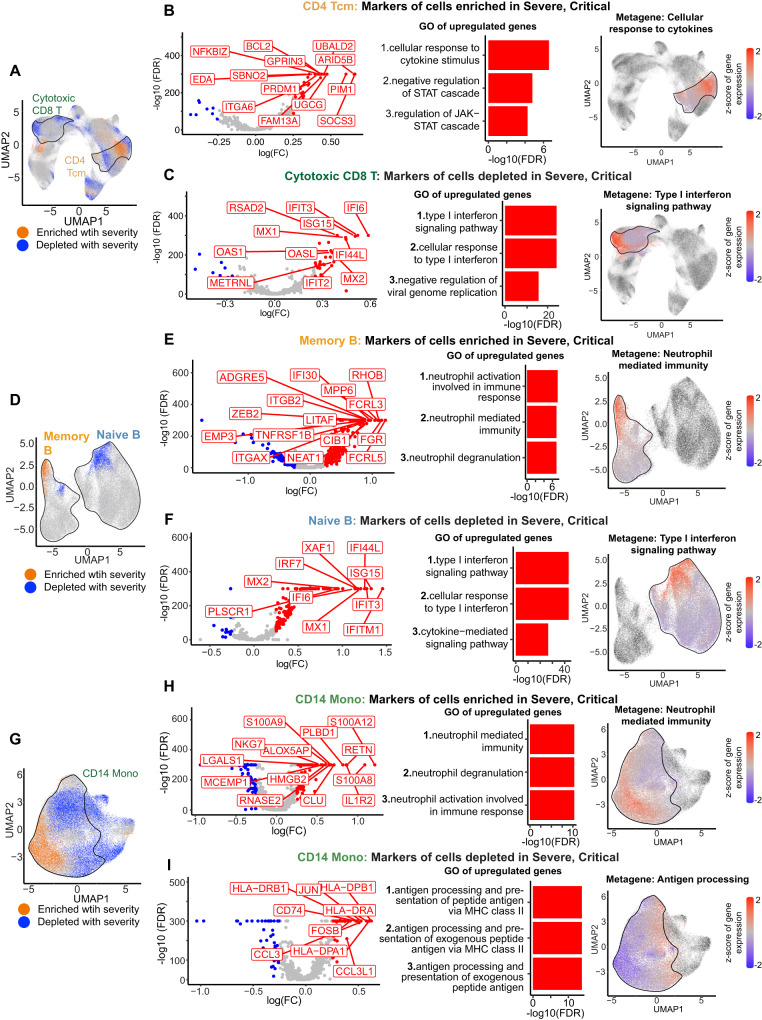
Due to the paucity of longitudinal molecular studies of COVID-19, particularly those covering the early stages of infection (Days 1-8 symptom onset), our understanding of host response over the disease course is limited. We perform longitudinal single cell RNA-seq on 286 blood samples from 108 age- and sex-matched COVID-19 patients, including 73 with early samples. We examine discrete cell subtypes and continuous cell states longitudinally, and we identify upregulation of type I IFN-stimulated genes (ISGs) as the predominant early signature of subsequent worsening of symptoms, which we validate in an independent cohort and corroborate by plasma markers. However, ISG expression is dynamic in progressors, spiking early and then rapidly receding to the level of severity-matched non-progressors. In contrast, cross-sectional analysis shows that ISG expression is deficient and IFN suppressors such as SOCS3 are upregulated in severe and critical COVID-19. We validate the latter in four independent cohorts, and SOCS3 inhibition reduces SARS-CoV-2 replication in vitro. In summary, we identify complexity in type I IFN response to COVID-19, as well as a potential avenue for host-directed therapy.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA323, 1239–1242 (2020). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
