

Temporal Dynamics, Discovery, and Emergence of Human-Transmissible RNA Viruses
- PMID: 38241079
- PMCID: PMC10797954
- DOI: 10.1093/molbev/msad272
Temporal Dynamics, Discovery, and Emergence of Human-Transmissible RNA Viruses
Abstract
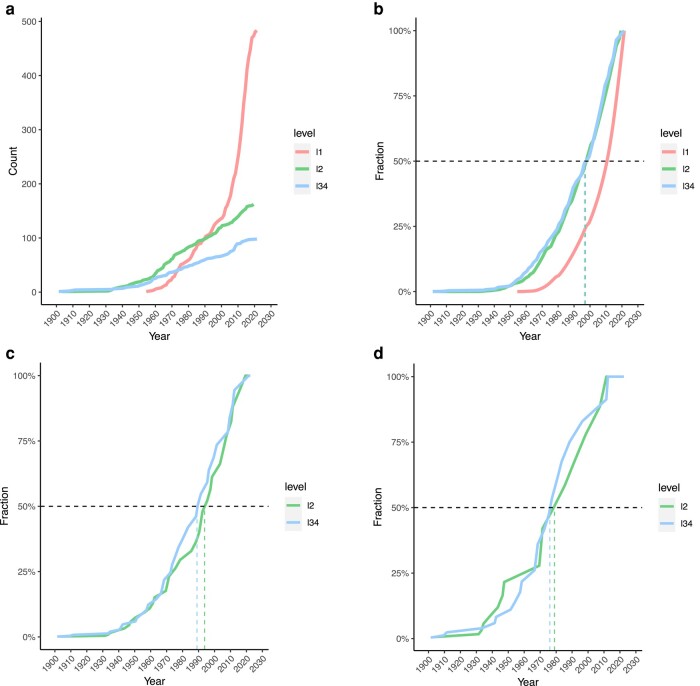
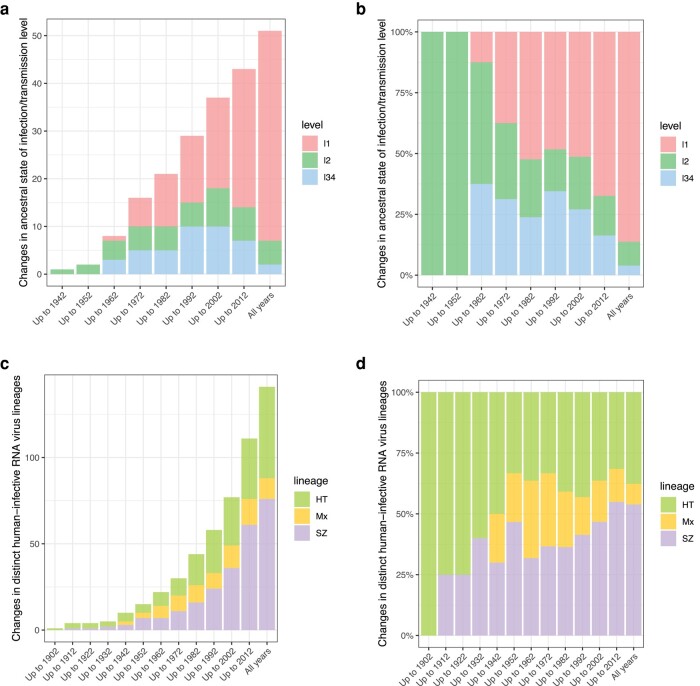
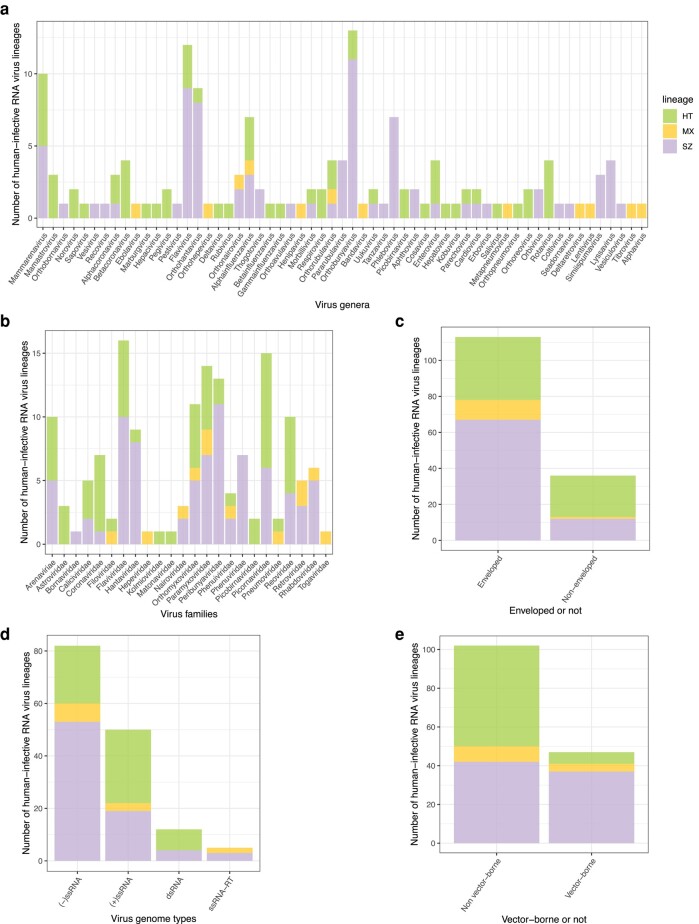
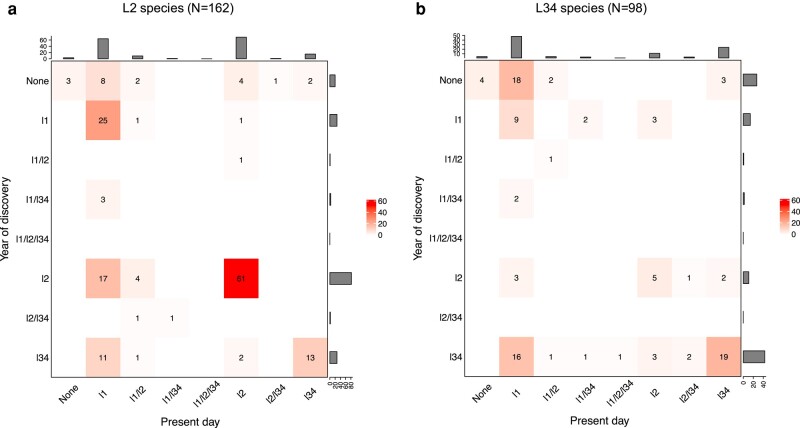
Transmissibility, the ability to spread within host populations, is a prerequisite for a pathogen to have epidemic or pandemic potential. Here, we estimate the phylogenies of human infectivity and transmissibility using 1,408 genome sequences from 743 distinct RNA virus species/types in 59 genera. By repeating this analysis using data sets censored by virus discovery date, we explore how temporal changes in the known diversity of RNA viruses-especially recent increases in recognized nonhuman viruses-have altered these phylogenies. Over time, we find significant increases in the proportion of RNA virus genera estimated to have a nonhuman-infective ancestral state, in the fraction of distinct human virus lineages that are purely human-transmissible or strictly zoonotic (compared to mixed lineages), and in the number of human viruses with nearest relatives known not to infect humans. Our results are consistent with viruses that are capable of spreading in human populations commonly emerging from a nonhuman reservoir. This is more likely in lineages that already contain human-transmissible viruses but is rare in lineages that contain only strictly zoonotic viruses.
Keywords: RNA viruses; emergences; epidemic potential; phylogenetics; temporal dynamics.
© The Author(s) 2024. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
