

Mitochondrial dysfunction in neurodegenerative disorders
- PMID: 38241161
- PMCID: PMC10903104
- DOI: 10.1016/j.neurot.2023.10.002
Mitochondrial dysfunction in neurodegenerative disorders
Abstract
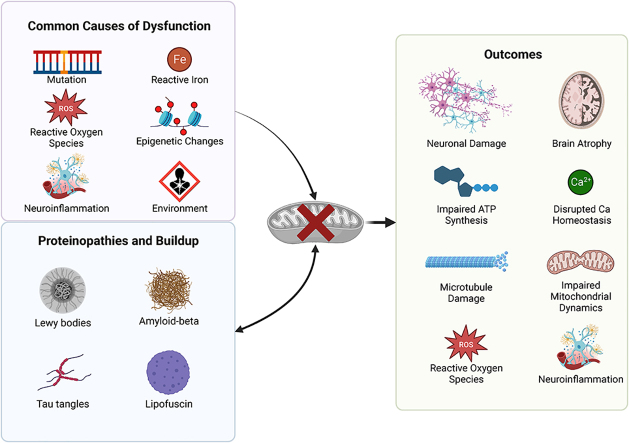
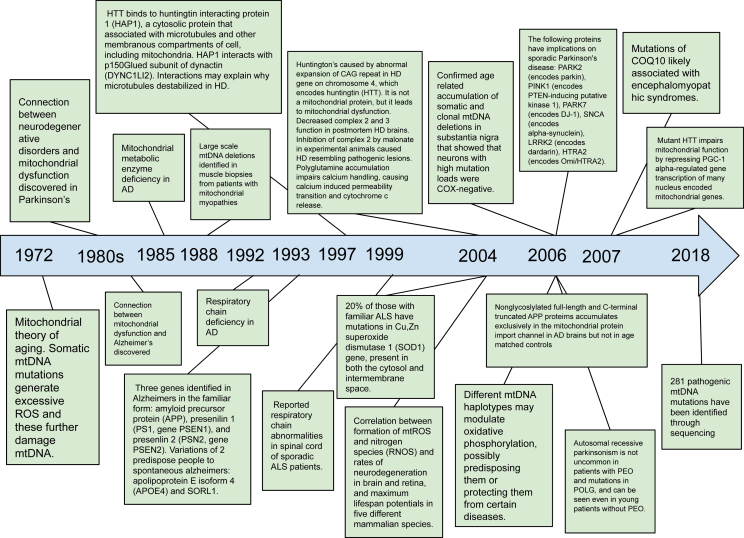
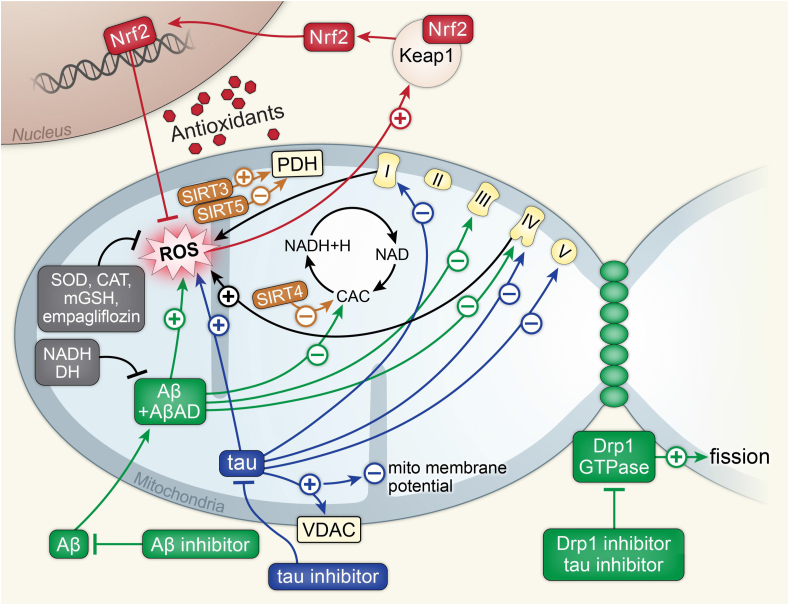
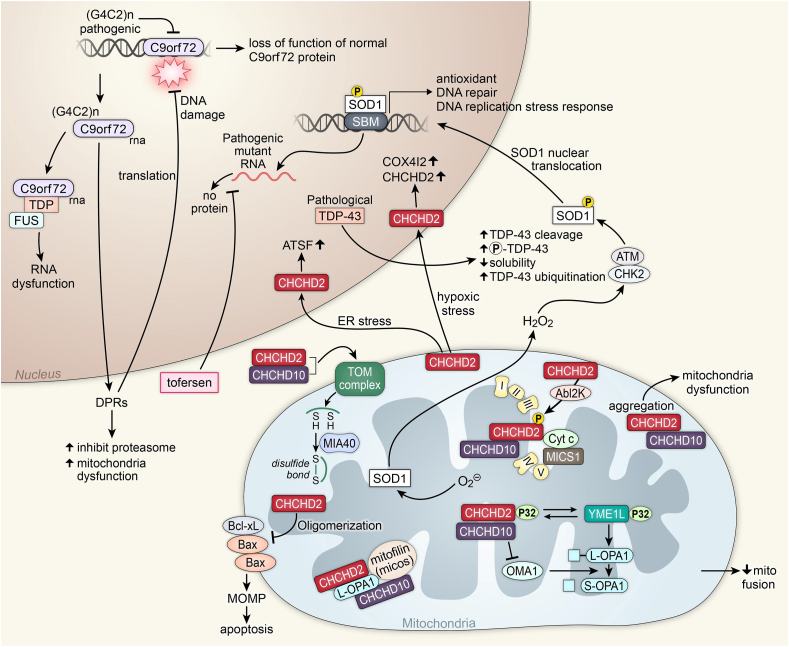
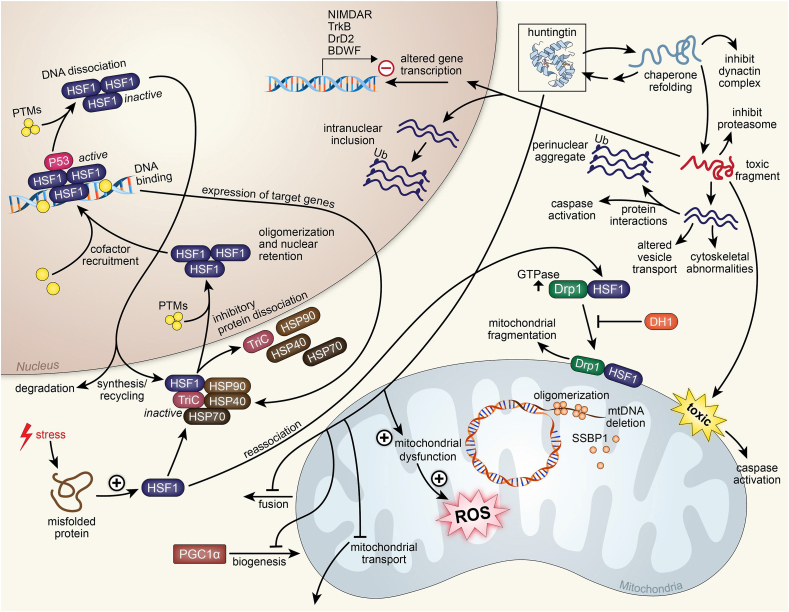
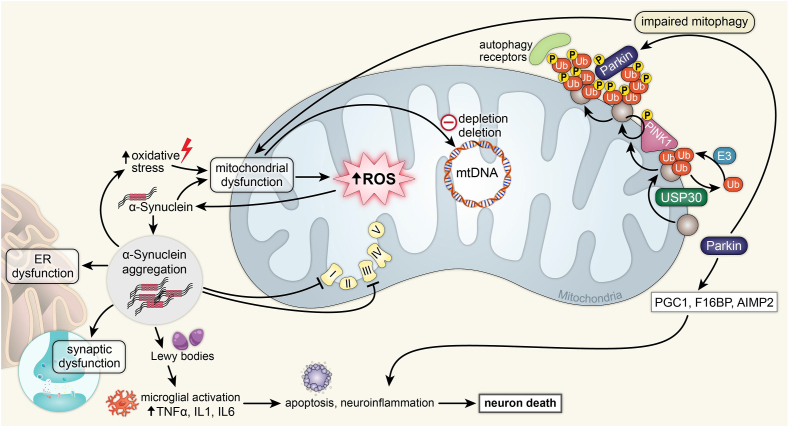
Recent advances in understanding the role of mitochondrial dysfunction in neurodegenerative diseases have expanded the opportunities for neurotherapeutics targeting mitochondria to alleviate symptoms and slow disease progression. In this review, we offer a historical account of advances in mitochondrial biology and neurodegenerative disease. Additionally, we summarize current knowledge of the normal physiology of mitochondria and the pathogenesis of mitochondrial dysfunction, the role of mitochondrial dysfunction in neurodegenerative disease, current therapeutics and recent therapeutic advances, as well as future directions for neurotherapeutics targeting mitochondrial function. A focus is placed on reactive oxygen species and their role in the disruption of telomeres and their effects on the epigenome. The effects of mitochondrial dysfunction in the etiology and progression of Alzheimer's disease, amyotrophic lateral sclerosis, Parkinson's disease, and Huntington's disease are discussed in depth. Current clinical trials for mitochondria-targeting neurotherapeutics are discussed.
Keywords: Aging; Bioenergetics; Mitochondria; Neurodegeneration; Reactive oxygen species (ROS).
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Ballard J.W., Whitlock M.C. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–744. - PubMed
-
- Di Donato S. Disorders related to mitochondrial membranes: pathology of the respiratory chain and neurodegeneration. J Inherit Metab Dis. 2000;23:247–263. - PubMed
-
- Frey T.G., Mannella C.A. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–324. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
