

Mitochondrial and metabolic dysfunction of peripheral immune cells in multiple sclerosis
- PMID: 38243312
- PMCID: PMC10799425
- DOI: 10.1186/s12974-024-03016-8
Mitochondrial and metabolic dysfunction of peripheral immune cells in multiple sclerosis
Abstract
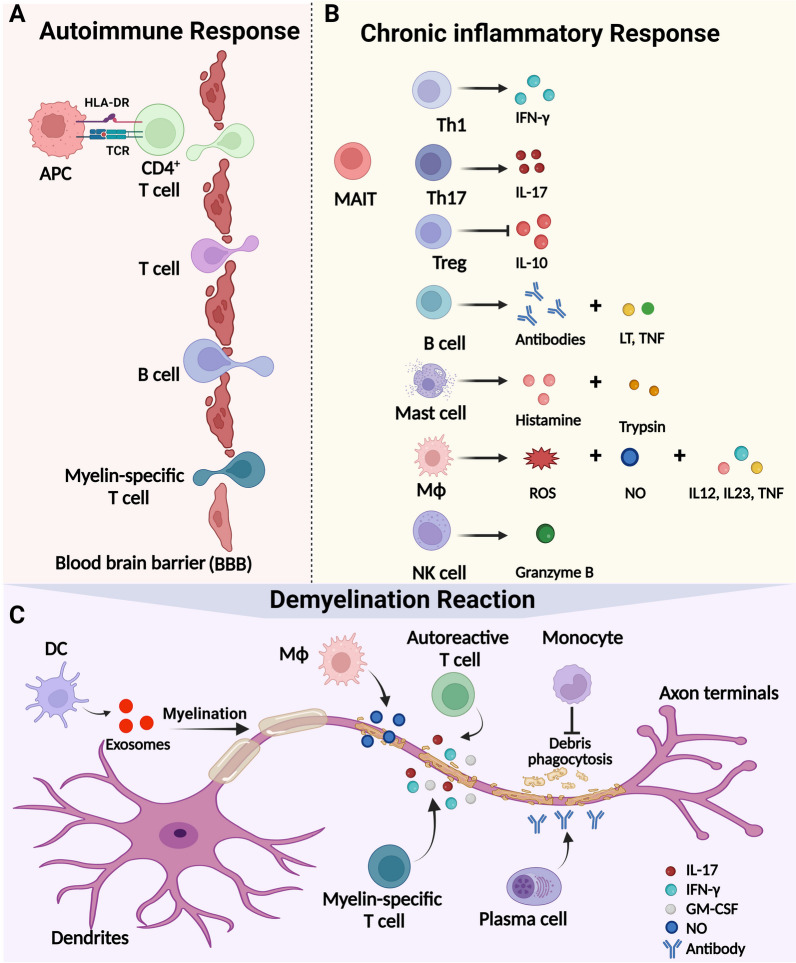
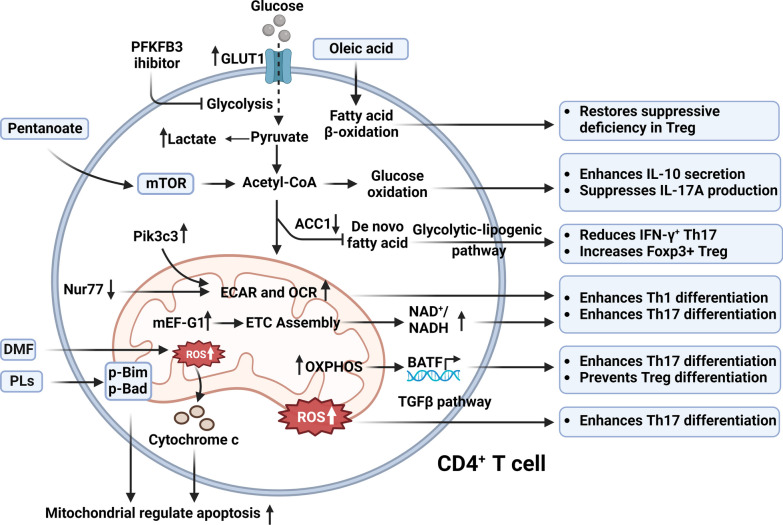
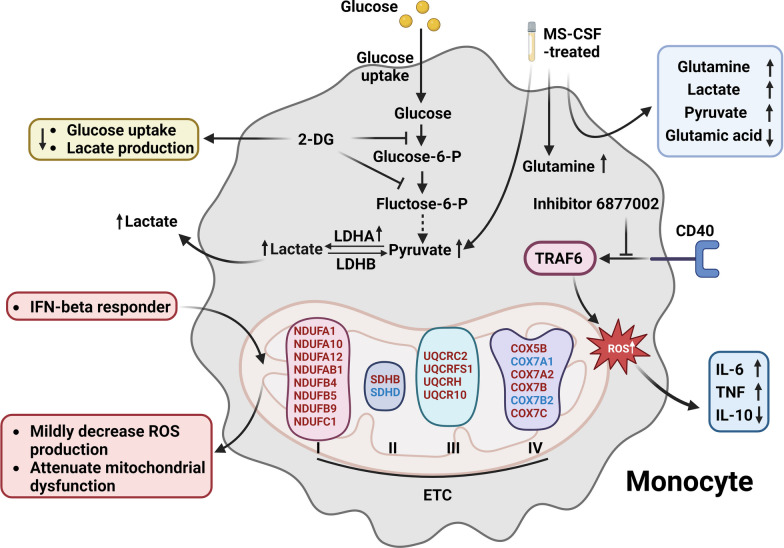
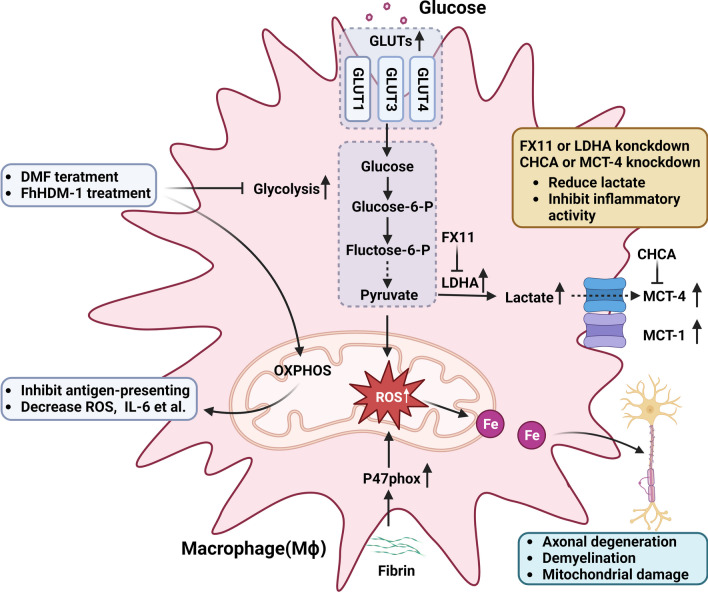
Multiple sclerosis (MS) is a chronic autoimmune disorder characterized by the infiltration of inflammatory cells and demyelination of nerves. Mitochondrial dysfunction has been implicated in the pathogenesis of MS, as studies have shown abnormalities in mitochondrial activities, metabolism, mitochondrial DNA (mtDNA) levels, and mitochondrial morphology in immune cells of individuals with MS. The presence of mitochondrial dysfunctions in immune cells contributes to immunological dysregulation and neurodegeneration in MS. This review provided a comprehensive overview of mitochondrial dysfunction in immune cells associated with MS, focusing on the potential consequences of mitochondrial metabolic reprogramming on immune function. Current challenges and future directions in the field of immune-metabolic MS and its potential as a therapeutic target were also discussed.
Keywords: Immune cells; Immune-metabolic; MS; Mitochondrion.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests. Figures were created with biorender.com.
Figures
Similar articles
-
The role of mitochondrial DNA variants and dysfunction in the pathogenesis and progression of multiple sclerosis.Mitochondrion. 2025 Mar;81:102002. doi: 10.1016/j.mito.2024.102002. Epub 2024 Dec 26. Mitochondrion. 2025. PMID: 39732186 Review.
-
Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis.Trends Mol Med. 2014 Mar;20(3):179-87. doi: 10.1016/j.molmed.2013.11.007. Epub 2013 Dec 24. Trends Mol Med. 2014. PMID: 24369898 Review.
-
Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis.FEBS Lett. 2018 Apr;592(7):1113-1121. doi: 10.1002/1873-3468.13013. Epub 2018 Mar 25. FEBS Lett. 2018. PMID: 29453889 Review.
-
Clonal expansion of mitochondrial DNA deletions and the progression of multiple sclerosis.CNS Neurol Disord Drug Targets. 2012 Aug;11(5):589-97. doi: 10.2174/187152712801661194. CNS Neurol Disord Drug Targets. 2012. PMID: 22583438 Review.
-
Mitochondria-targeted Antioxidants as a Prospective Therapeutic Strategy for Multiple Sclerosis.Curr Med Chem. 2017;24(19):2086-2114. doi: 10.2174/0929867324666170316114452. Curr Med Chem. 2017. PMID: 28302008
Cited by
-
Modulatory Effects of Urtica dioica on Neurodegenerative Diseases: Unveiling the Latest Findings and Applications Related to Neuroinflammation, Oxidative Stress, and Cognitive Dysfunction.Antioxidants (Basel). 2025 Jul 12;14(7):854. doi: 10.3390/antiox14070854. Antioxidants (Basel). 2025. PMID: 40722958 Free PMC article. Review.
-
Impact of Epstein-Barr Virus Nuclear Antigen 1 on Neuroinflammation in PARK2 Knockout Mice.Int J Mol Sci. 2024 Oct 4;25(19):10697. doi: 10.3390/ijms251910697. Int J Mol Sci. 2024. PMID: 39409029 Free PMC article.
-
Oxidized Low-Density Lipoprotein and Its Role in Immunometabolism.Int J Mol Sci. 2024 Oct 23;25(21):11386. doi: 10.3390/ijms252111386. Int J Mol Sci. 2024. PMID: 39518939 Free PMC article. Review.
-
An inflammation-associated lncRNA induces neuronal damage via mitochondrial dysfunction.Mol Ther Nucleic Acids. 2025 Apr 2;36(2):102533. doi: 10.1016/j.omtn.2025.102533. eCollection 2025 Jun 10. Mol Ther Nucleic Acids. 2025. PMID: 40291376 Free PMC article.
-
The Connection Between Oxidative Stress, Mitochondrial Dysfunction, Iron Metabolism and Microglia in Multiple Sclerosis: A Narrative Review.NeuroSci. 2025 Mar 4;6(1):23. doi: 10.3390/neurosci6010023. NeuroSci. 2025. PMID: 40137866 Free PMC article. Review.
References
-
- Jakimovski D, Bittner S, Zivadinov R, Morrow SA, Benedict RH, Zipp F, Weinstock-Guttman B. Multiple sclerosis. Lancet. 2023. - PubMed
-
- Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, Robertson N, La Rocca N, Uitdehaag B, van der Mei I, Wallin M, Helme A, Angood Napier C, Rijke N, Baneke P. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult Scler. 2020;26:1816–1821. doi: 10.1177/1352458520970841. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
