

Modeling the limits of detection for antimicrobial resistance genes in agri-food samples: a comparative analysis of bioinformatics tools
- PMID: 38245666
- PMCID: PMC10799530
- DOI: 10.1186/s12866-023-03148-6
Modeling the limits of detection for antimicrobial resistance genes in agri-food samples: a comparative analysis of bioinformatics tools
Abstract
Background: Although the spread of antimicrobial resistance (AMR) through food and its production poses a significant concern, there is limited research on the prevalence of AMR bacteria in various agri-food products. Sequencing technologies are increasingly being used to track the spread of AMR genes (ARGs) in bacteria, and metagenomics has the potential to bypass some of the limitations of single isolate characterization by allowing simultaneous analysis of the agri-food product microbiome and associated resistome. However, metagenomics may still be hindered by methodological biases, presence of eukaryotic DNA, and difficulties in detecting low abundance targets within an attainable sequence coverage. The goal of this study was to assess whether limits of detection of ARGs in agri-food metagenomes were influenced by sample type and bioinformatic approaches.
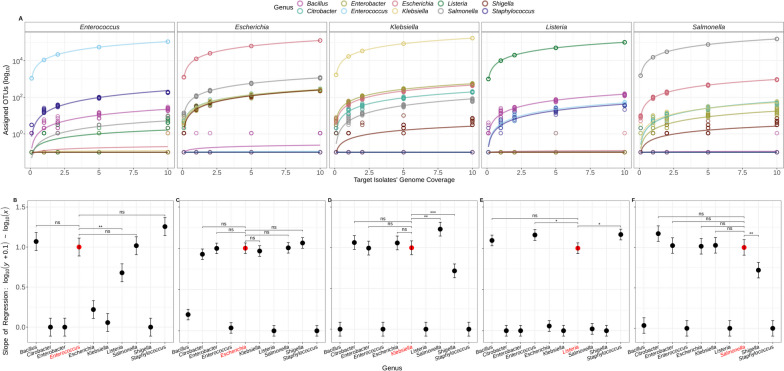
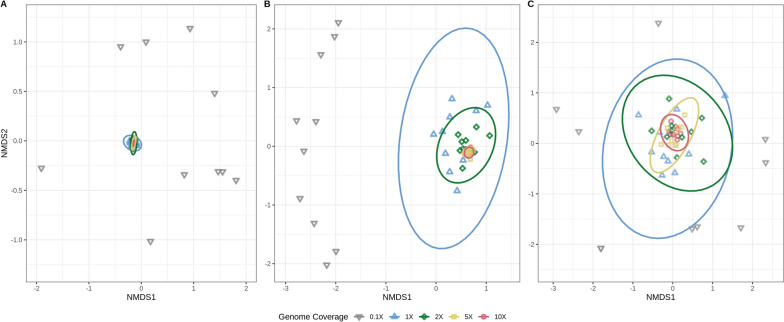
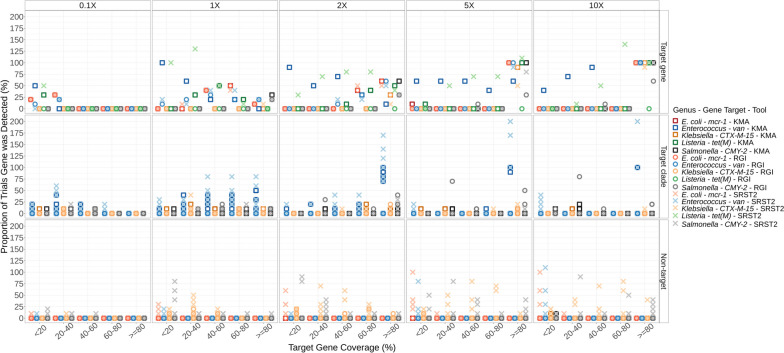
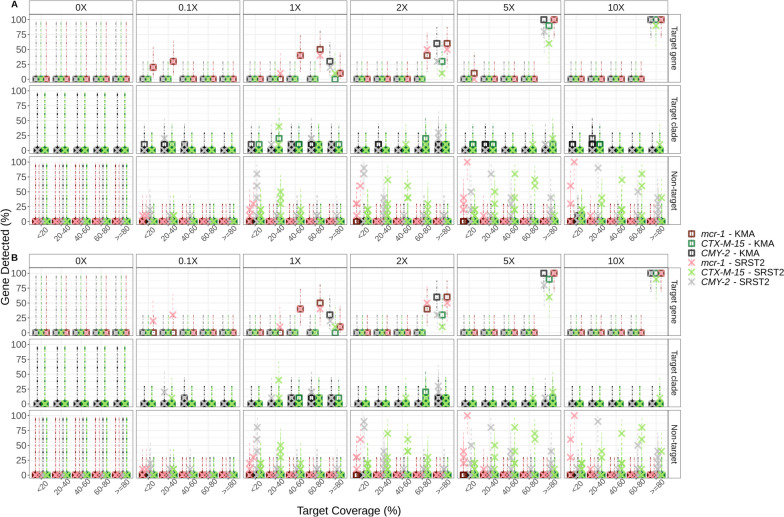
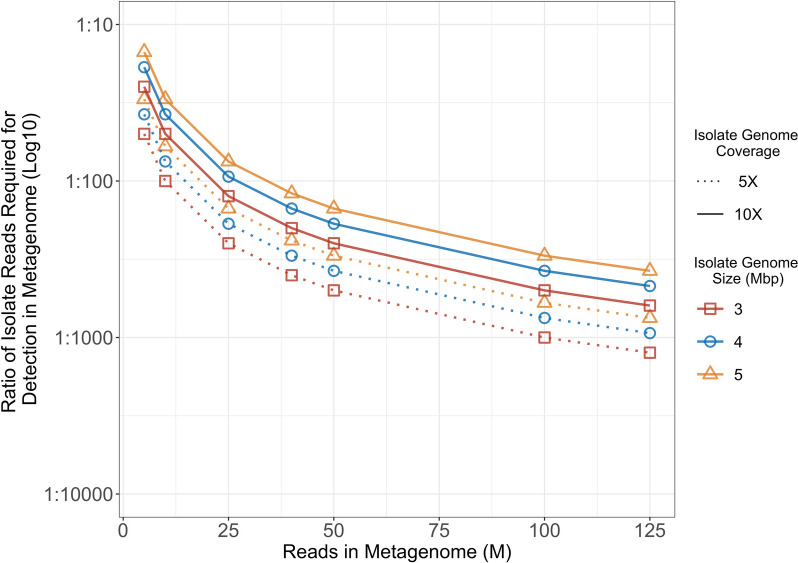
Results: We simulated metagenomes containing different proportions of AMR pathogens and analysed them for taxonomic composition and ARGs using several common bioinformatic tools. Kraken2/Bracken estimates of species abundance were closest to expected values. However, analysis by both Kraken2/Bracken indicated presence of organisms not included in the synthetic metagenomes. Metaphlan3/Metaphlan4 analysis of community composition was more specific but with lower sensitivity than the Kraken2/Bracken analysis. Accurate detection of ARGs dropped drastically below 5X isolate genome coverage. However, it was sometimes possible to detect ARGs and closely related alleles at lower coverage levels if using a lower ARG-target coverage cutoff (< 80%). While KMA and CARD-RGI only predicted presence of expected ARG-targets or closely related gene-alleles, SRST2 (which allows read to map to multiple targets) falsely reported presence of distantly related ARGs at all isolate genome coverage levels. The presence of background microbiota in metagenomes influenced the accuracy of ARG detection by KMA, resulting in mcr-1 detection at 0.1X isolate coverage in the lettuce but not in the beef metagenome.
Conclusions: This study demonstrates accurate detection of ARGs in synthetic metagenomes using various bioinformatic methods, provided that reads from the ARG-encoding organism exceed approximately 5X isolate coverage (i.e. 0.4% of a 40 million read metagenome). While lowering thresholds for target gene detection improved sensitivity, this led to the identification of alternative ARG-alleles, potentially confounding the identification of critical ARGs in the resistome. Further advancements in sequencing technologies providing increased coverage depth or extended read lengths may improve ARG detection in agri-food metagenomic samples, enabling use of this approach for tracking clinically important ARGs in agri-food samples.
Keywords: Antimicrobial resistance; Limit of detection; Metagenomics; Sequence coverage.
© 2024. Crown.
Conflict of interest statement
Competing interests. ST (Sandeep Tamber) is a member of the BMC Microbiology Editorial board.We have no other competing interests. The authors declare that they have no other competing interests.
Figures

References
-
- Bengtsson-Palme J. Antibiotic resistance in the food supply chain: where can sequencing and metagenomics aid risk assessment? Curr Opin Food Sci. 2017;1(14):66–71. doi: 10.1016/j.cofs.2017.01.010. - DOI
-
- Hudson JA, Frewer LJ, Jones G, Brereton PA, Whittingham MJ, Stewart G. The agri-food chain and antimicrobial resistance: a review. Trends Food Sci Technol. 2017;1(69):131–147. doi: 10.1016/j.tifs.2017.09.007. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
