

DLG3 variants caused X-linked epilepsy with/without neurodevelopmental disorders and the genotype-phenotype correlation
- PMID: 38249294
- PMCID: PMC10796462
- DOI: 10.3389/fnmol.2023.1290919
DLG3 variants caused X-linked epilepsy with/without neurodevelopmental disorders and the genotype-phenotype correlation
Abstract
Background: The DLG3 gene encodes disks large membrane-associated guanylate kinase scaffold protein 3, which plays essential roles in the clustering of N-methyl-D-aspartate receptors (NMDARs) at excitatory synapses. Previously, DLG3 has been identified as the causative gene of X-linked intellectual developmental disorder-90 (XLID-90; OMIM# 300850). This study aims to explore the phenotypic spectrum of DLG3 and the genotype-phenotype correlation.
Methods: Trios-based whole-exome sequencing was performed in patients with epilepsy of unknown causes. To analyze the genotype-phenotype correlations, previously reported DLG3 variants were systematically reviewed.
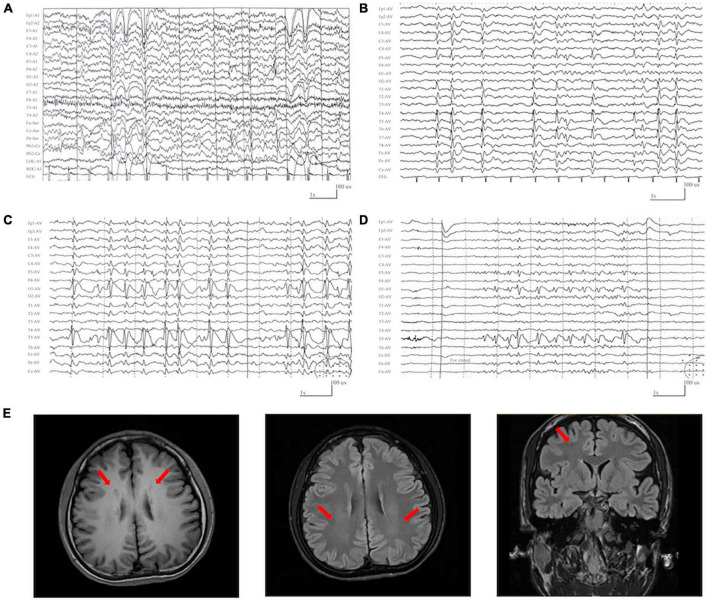
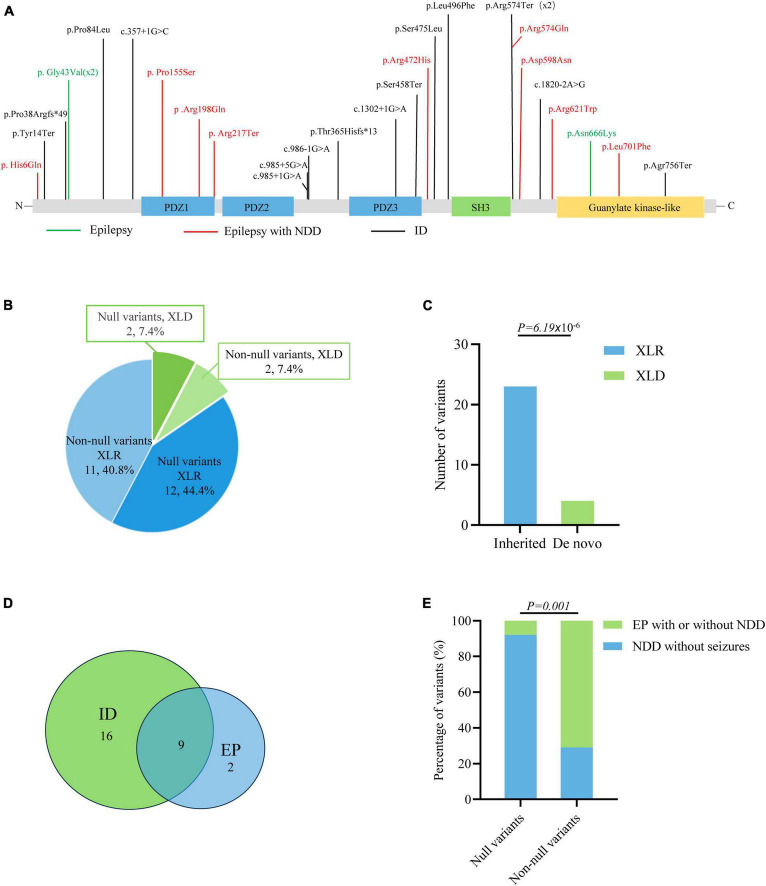
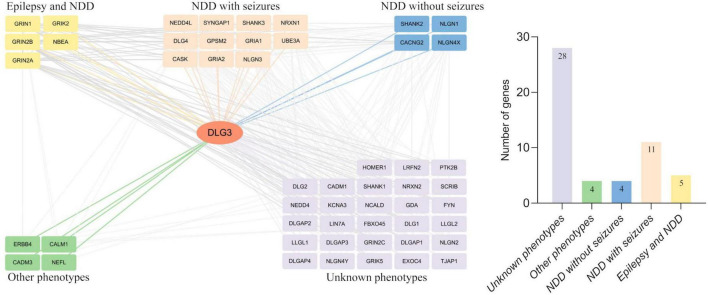
Results: DLG3 variants were identified in seven unrelated cases with epilepsy. These variants had no hemizygous frequencies in controls. All variants were predicted to be damaging by silico tools and alter the hydrogen bonds with surrounding residues and/or protein stability. Four cases mainly presented with generalized seizures, including generalized tonic-clonic and myoclonic seizures, and the other three cases exhibited secondary generalized tonic-clonic seizures and focal seizures. Multifocal discharges were recorded in all cases during electroencephalography monitoring, including the four cases with generalized discharges initially but multifocal discharges after drug treating. Protein-protein interaction network analysis revealed that DLG3 interacts with 52 genes with high confidence, in which the majority of disease-causing genes were associated with a wide spectrum of neurodevelopmental disorder (NDD) and epilepsy. Three patients with variants locating outside functional domains all achieved seizure-free, while the four patients with variants locating in functional domains presented poor control of seizures. Analysis of previously reported cases revealed that patients with non-null variants presented higher percentages of epilepsy than those with null variants, suggesting a genotype-phenotype correlation.
Significance: This study suggested that DLG3 variants were associated with epilepsy with/without NDD, expanding the phenotypic spectrum of DLG3. The observed genotype-phenotype correlation potentially contributes to the understanding of the underlying mechanisms driving phenotypic variation.
Keywords: DLG3 gene; Genotype-phenotype correlation; epilepsy; neurodevelopmental disorder; variants.
Copyright © 2024 He, Luo, Jin, Wang, Xu, Jiao, Yan, Wang, Zhai, Ji, Zhang, Zhou, Li, Liao, Lan and Xu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures




References
LinkOut - more resources
Full Text Sources