

Unraveling the Behavior of Intrinsically Disordered Protein c-Myc: A Study Utilizing Gaussian-Accelerated Molecular Dynamics
- PMID: 38250404
- PMCID: PMC10795134
- DOI: 10.1021/acsomega.3c05822
Unraveling the Behavior of Intrinsically Disordered Protein c-Myc: A Study Utilizing Gaussian-Accelerated Molecular Dynamics
Abstract
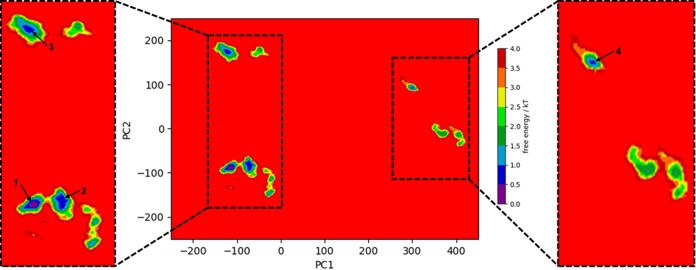
The protein c-Myc is a transcription factor that remains largely intrinsically disordered and is known to be involved in various biological processes and is overexpressed in various cancers, making it an attractive drug target. However, intrinsically disordered proteins such as c-Myc do not show funnel-like basins in their free-energy landscapes; this makes their druggability a challenge. For the first time, we propose a heterodimer model of c-Myc/Max in full length in this work. We used Gaussian-accelerated molecular dynamics (GaMD) simulations to explore the behavior of c-Myc and its various regions, including the transactivation domain (TAD) and the basic helix-loop-helix-leucine-zipper (bHLH-Zipper) motif in three different conformational states: (a) monomeric c-Myc, (b) c-Myc when bound to its partner protein, Max, and (c) when Max was removed after binding. We analyzed the GaMD trajectories using root-mean-square deviation (RMSD), radius of gyration, root-mean-square fluctuation, and free-energy landscape (FEL) calculations to elaborate the behaviors of these regions. The results showed that the monomeric c-Myc structure showed a higher RMSD fluctuation as compared with the c-Myc/Max heterodimer in the bHLH-Zipper motif. This indicated that the bHLH-Zipper motif of c-Myc is more stable when it is bound to Max. The TAD region in both monomeric and Max-bound states showed similar plasticity in terms of RMSD. We also conducted residue decomposition calculations and showed that the c-Myc and Max interaction could be driven mainly by electrostatic interactions and the residues Arg299, Ile403, and Leu420 seemed to play important roles in the interaction. Our work provides insights into the behavior of c-Myc and its regions that could support the development of drugs that target c-Myc and other intrinsically disordered proteins.
© 2023 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures










Similar articles
-
Myc phosphorylation in its basic helix-loop-helix region destabilizes transient α-helical structures, disrupting Max and DNA binding.J Biol Chem. 2018 Jun 15;293(24):9301-9310. doi: 10.1074/jbc.RA118.002709. Epub 2018 Apr 25. J Biol Chem. 2018. PMID: 29695509 Free PMC article.
-
The Disordered MAX N-terminus Modulates DNA Binding of the Transcription Factor MYC:MAX.J Mol Biol. 2022 Nov 30;434(22):167833. doi: 10.1016/j.jmb.2022.167833. Epub 2022 Sep 27. J Mol Biol. 2022. PMID: 36174765
-
Intrinsically Disordered Regions in the Transcription Factor MYC:MAX Modulate DNA Binding via Intramolecular Interactions.Biochemistry. 2024 Jan 24. doi: 10.1021/acs.biochem.3c00608. Online ahead of print. Biochemistry. 2024. PMID: 38264995
-
Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein.Cells. 2020 Apr 22;9(4):1038. doi: 10.3390/cells9041038. Cells. 2020. PMID: 32331235 Free PMC article. Review.
-
The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation.Oncogene. 1999 May 13;18(19):2955-66. doi: 10.1038/sj.onc.1202750. Oncogene. 1999. PMID: 10378692 Review.
Cited by
-
Current perspectives in drug targeting intrinsically disordered proteins and biomolecular condensates.BMC Biol. 2025 May 6;23(1):118. doi: 10.1186/s12915-025-02214-x. BMC Biol. 2025. PMID: 40325419 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources