

Deciphering cell states and genealogies of human haematopoiesis
- PMID: 38253266
- PMCID: PMC10937407
- DOI: 10.1038/s41586-024-07066-z
Deciphering cell states and genealogies of human haematopoiesis
Abstract
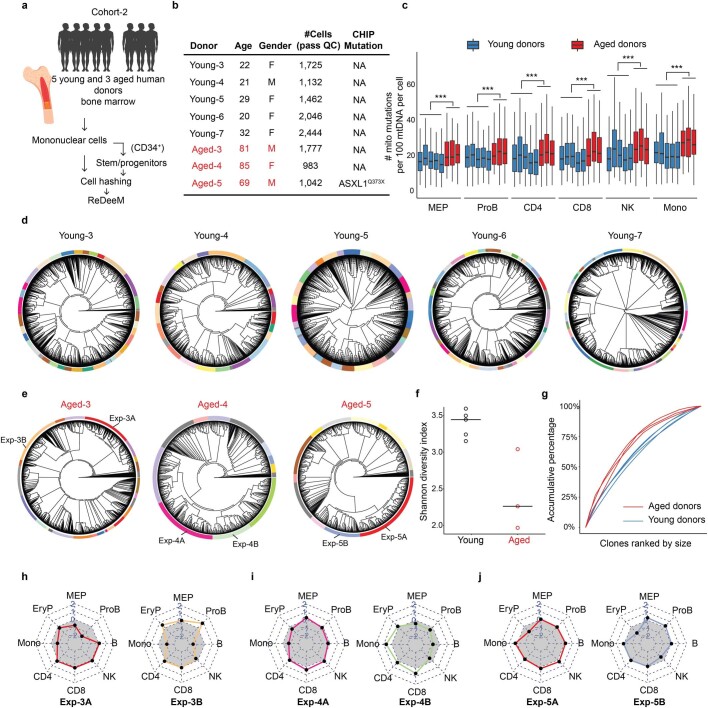
The human blood system is maintained through the differentiation and massive amplification of a limited number of long-lived haematopoietic stem cells (HSCs)1. Perturbations to this process underlie diverse diseases, but the clonal contributions to human haematopoiesis and how this changes with age remain incompletely understood. Although recent insights have emerged from barcoding studies in model systems2-5, simultaneous detection of cell states and phylogenies from natural barcodes in humans remains challenging. Here we introduce an improved, single-cell lineage-tracing system based on deep detection of naturally occurring mitochondrial DNA mutations with simultaneous readout of transcriptional states and chromatin accessibility. We use this system to define the clonal architecture of HSCs and map the physiological state and output of clones. We uncover functional heterogeneity in HSC clones, which is stable over months and manifests as both differences in total HSC output and biases towards the production of different mature cell types. We also find that the diversity of HSC clones decreases markedly with age, leading to an oligoclonal structure with multiple distinct clonal expansions. Our study thus provides a clonally resolved and cell-state-aware atlas of human haematopoiesis at single-cell resolution, showing an unappreciated functional diversity of human HSC clones and, more broadly, paving the way for refined studies of clonal dynamics across a range of tissues in human health and disease.
© 2024. The Author(s).
Conflict of interest statement
C.W., J.S.W. and V.G.S. are listed as inventors on a patent application covering the ReDeeM method. V.G.S. is an inventor on PCT/US2019/036583 covering the application of lineage tracing with mitochondrial genome mutations. V.G.S. serves as an advisor to and/or has equity in Branch Biosciences, Ensoma, Novartis and Cellarity. J.S.W. serves as an advisor to and/or has equity in 5 AM Ventures, Amgen, Chroma Medicine, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics and Tessera Therapeutics. The other authors declare no competing interests.
Figures






References
-
- Busch, K. et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature518, 542–546 (2015). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
