

Mapping three-dimensional intratumor proteomic heterogeneity in uterine serous carcinoma by multiregion microsampling
- PMID: 38254014
- PMCID: PMC10804562
- DOI: 10.1186/s12014-024-09451-2
Mapping three-dimensional intratumor proteomic heterogeneity in uterine serous carcinoma by multiregion microsampling
Abstract
Background: Although uterine serous carcinoma (USC) represents a small proportion of all uterine cancer cases, patients with this aggressive subtype typically have high rates of chemotherapy resistance and disease recurrence that collectively result in a disproportionately high death rate. The goal of this study was to provide a deeper view of the tumor microenvironment of this poorly characterized uterine cancer variant through multi-region microsampling and quantitative proteomics.
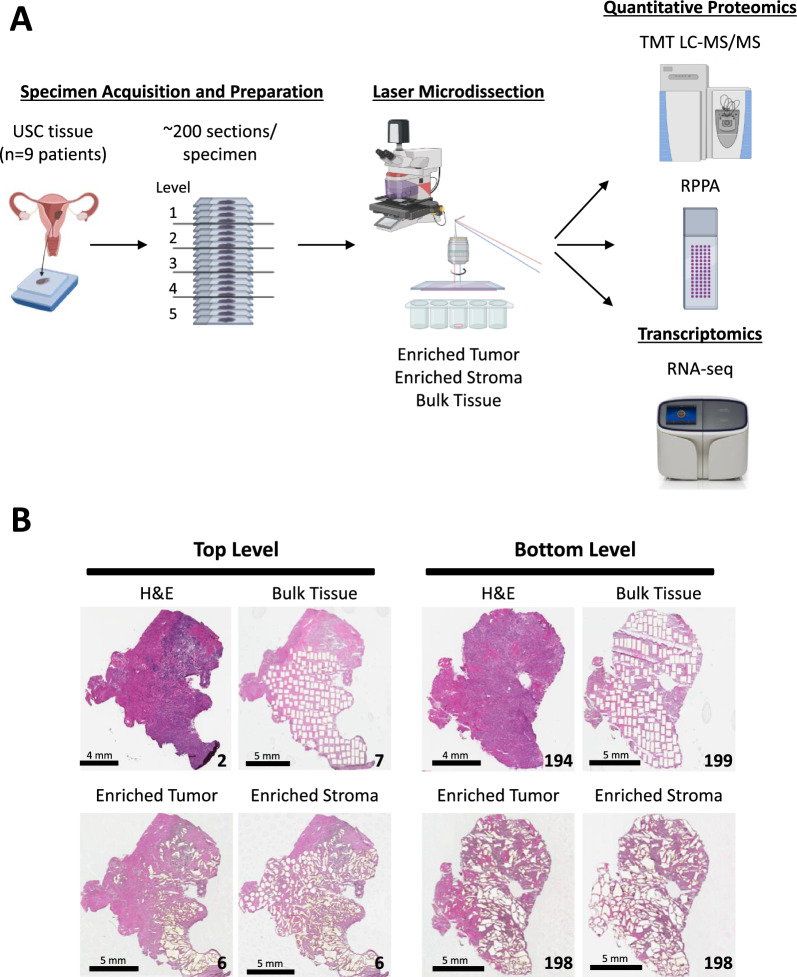
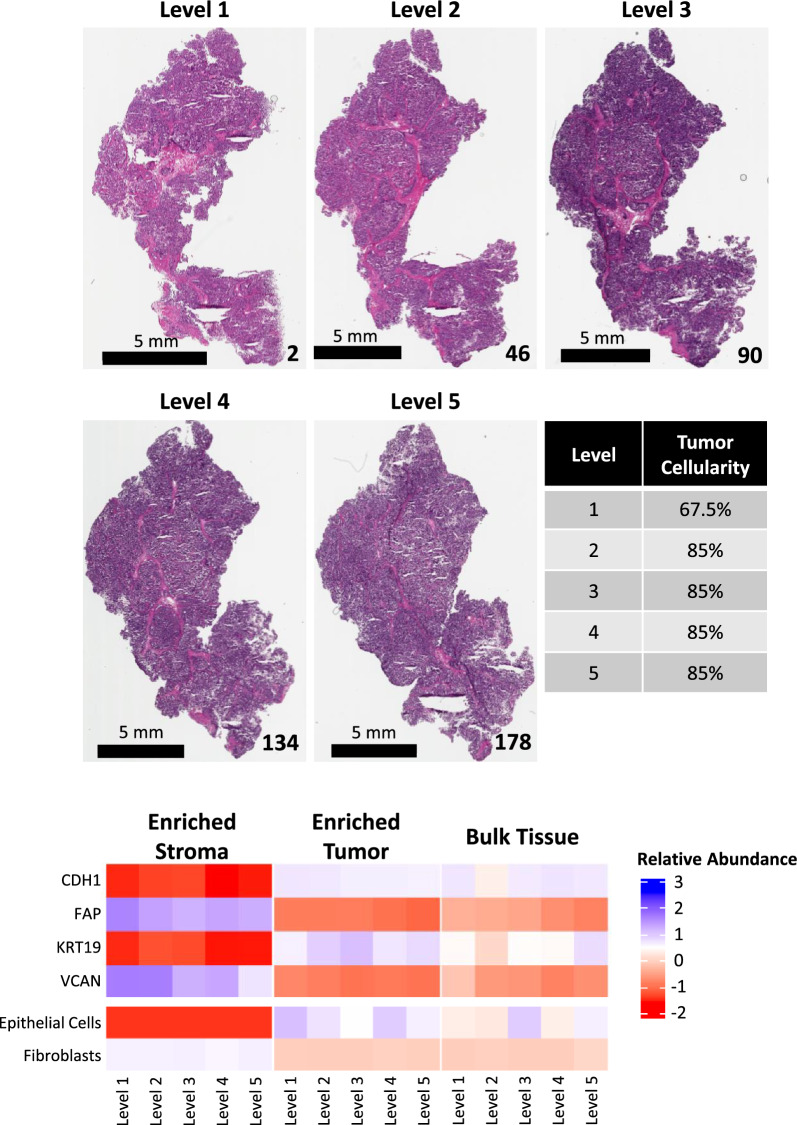
Methods: Tumor epithelium, tumor-involved stroma, and whole "bulk" tissue were harvested by laser microdissection (LMD) from spatially resolved levels from nine USC patient tumor specimens and underwent proteomic analysis by mass spectrometry and reverse phase protein arrays, as well as transcriptomic analysis by RNA-sequencing for one patient's tumor.
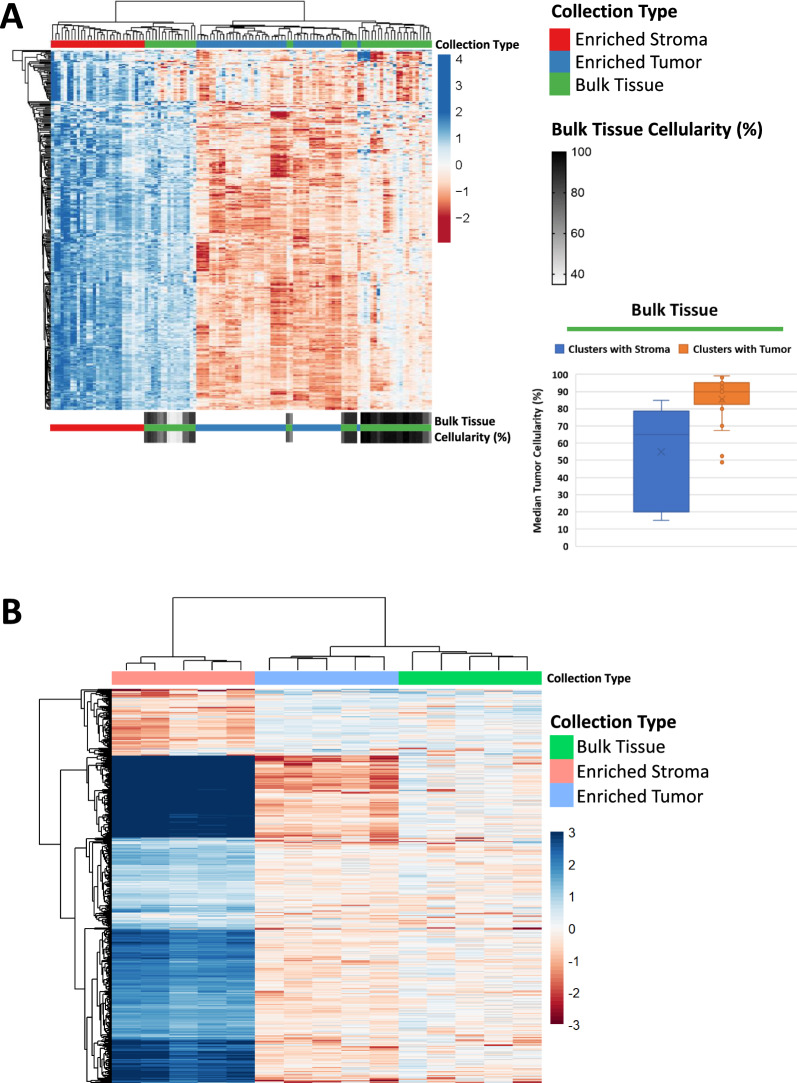
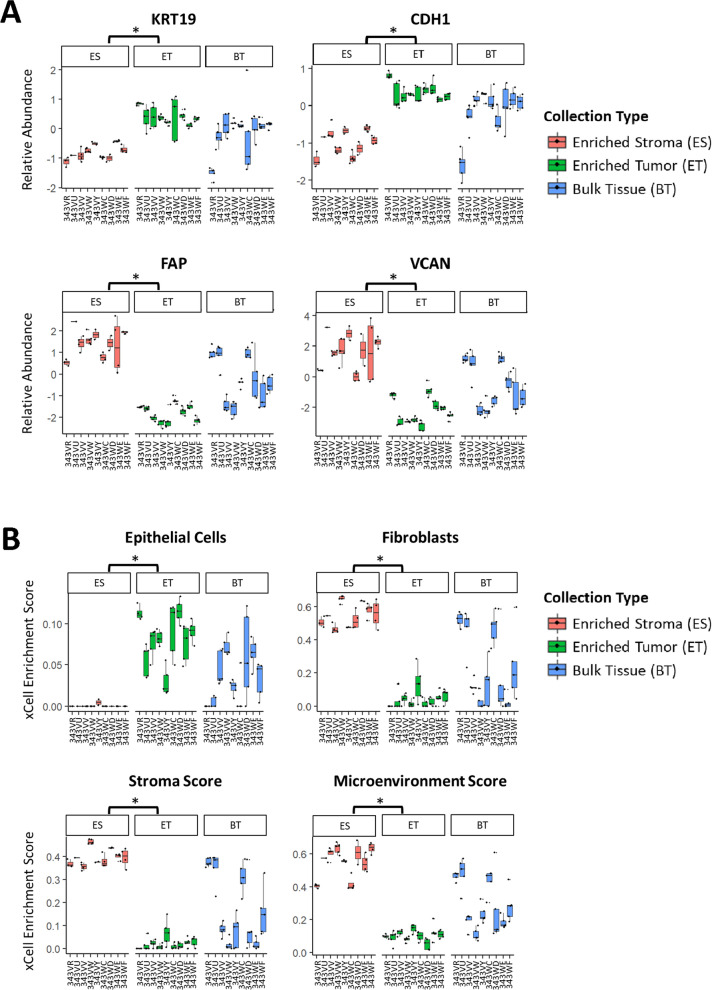
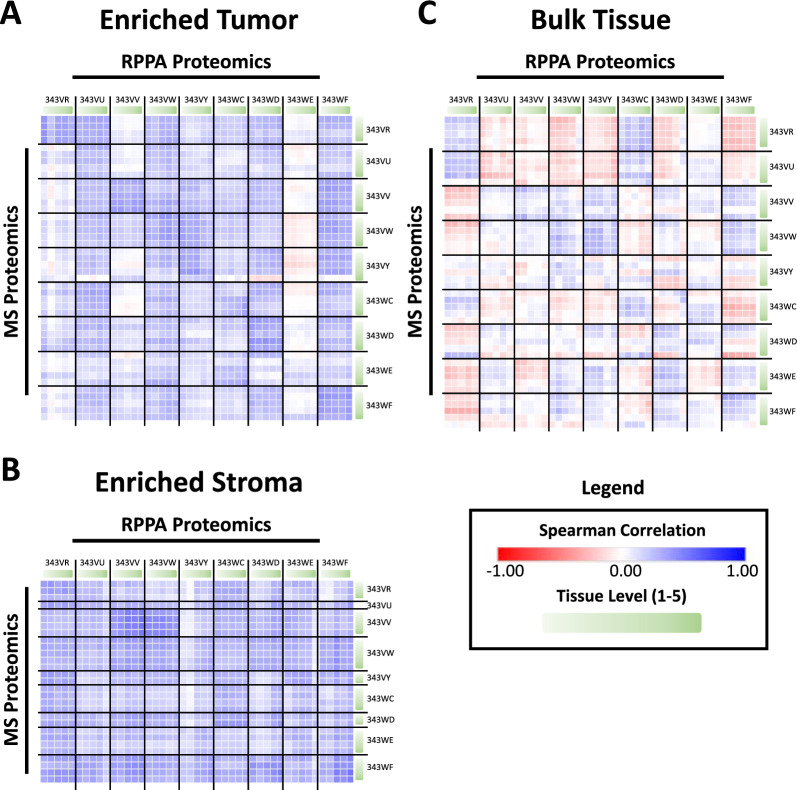
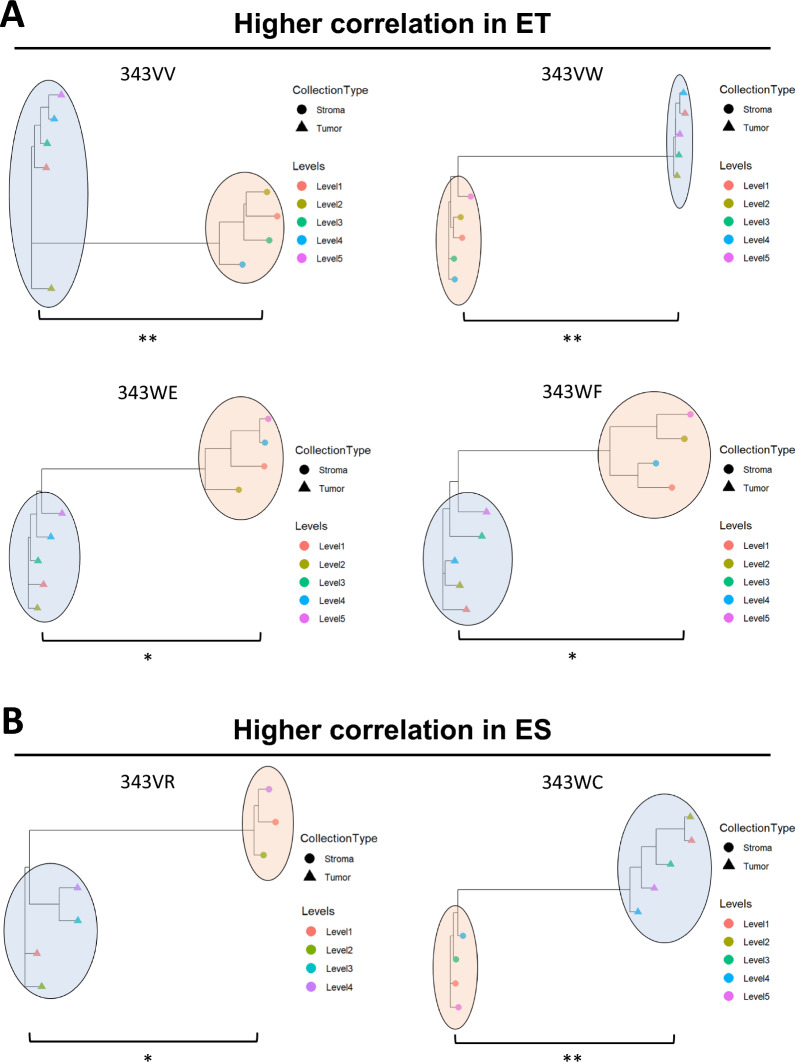
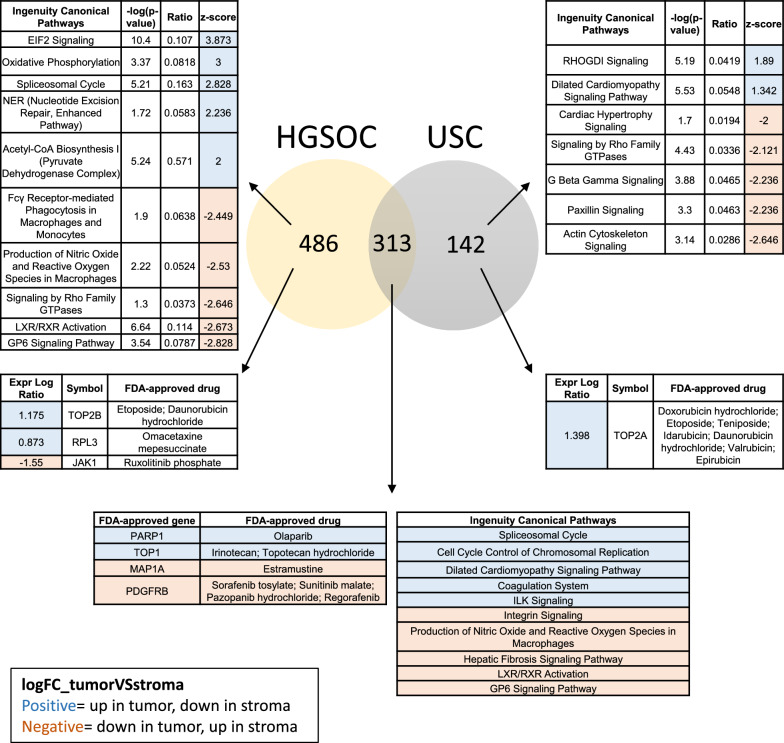
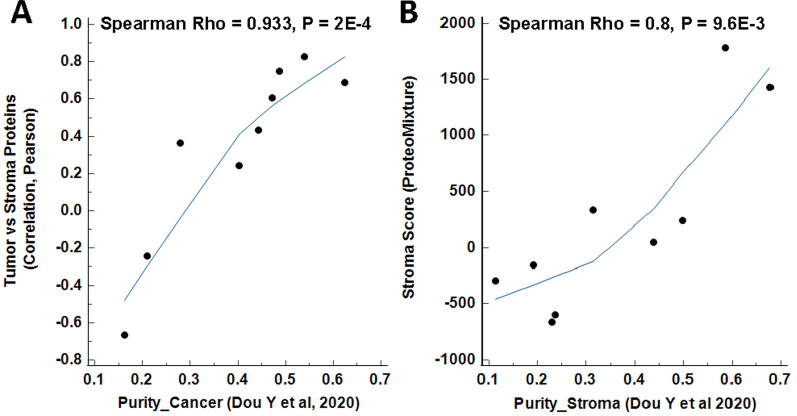
Results: LMD enriched cell subpopulations demonstrated varying degrees of relatedness, indicating substantial intratumor heterogeneity emphasizing the necessity for enrichment of cellular subpopulations prior to molecular analysis. Known prognostic biomarkers were quantified with stable levels in both LMD enriched tumor and stroma, which were shown to be highly variable in bulk tissue. These USC data were further used in a comparative analysis with a data generated from another serous gynecologic malignancy, high grade serous ovarian carcinoma, and have been added to our publicly available data analysis tool, the Heterogeneity Analysis Portal ( https://lmdomics.org/ ).
Conclusions: Here we identified extensive three-dimensional heterogeneity within the USC tumor microenvironment, with disease-relevant biomarkers present in both the tumor and the stroma. These data underscore the critical need for upfront enrichment of cellular subpopulations from tissue specimens for spatial proteogenomic analysis.
Keywords: Intratumor heterogeneity; Laser microdissection; Proteogenomics; Proteomics; Spatial proteomics; Tumor microenvironment; Uterine serous carcinoma.
© 2024. The Author(s).
Conflict of interest statement
TPC is a ThermoFisher Scientific, Inc SAB member and receives research funding from AbbVie. EFP receives research funding from Genentech, Pfizer, AbbVie, and is a co-inventor of the RPPA technology described herein and receives royalties on the related license agreements.
Figures
Similar articles
-
Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens.iScience. 2021 Jun 21;24(7):102757. doi: 10.1016/j.isci.2021.102757. eCollection 2021 Jul 23. iScience. 2021. PMID: 34278265 Free PMC article.
-
ProteoMixture: A cell type deconvolution tool for bulk tissue proteomic data.iScience. 2024 Feb 12;27(3):109198. doi: 10.1016/j.isci.2024.109198. eCollection 2024 Mar 15. iScience. 2024. PMID: 38439970 Free PMC article.
-
Assessment of the differences in oncologic outcomes between patients with high-grade serous ovarian carcinoma and uterine serous carcinoma.J Obstet Gynaecol Res. 2024 Jan;50(1):86-94. doi: 10.1111/jog.15814. Epub 2023 Oct 19. J Obstet Gynaecol Res. 2024. PMID: 37854000
-
HER2 in uterine serous carcinoma: Current state and clinical perspectives.Am J Clin Pathol. 2023 Oct 3;160(4):341-351. doi: 10.1093/ajcp/aqad056. Am J Clin Pathol. 2023. PMID: 37267036 Review.
-
Linking uterine serous carcinoma to BRCA1/2-associated cancer syndrome: A meta-analysis and case report.Eur J Cancer. 2017 Feb;72:215-225. doi: 10.1016/j.ejca.2016.11.028. Epub 2016 Dec 31. Eur J Cancer. 2017. PMID: 28049106 Review.
Cited by
-
Proteogenomics in Nephrology: A New Frontier in Nephrological Research.Curr Issues Mol Biol. 2024 May 11;46(5):4595-4608. doi: 10.3390/cimb46050279. Curr Issues Mol Biol. 2024. PMID: 38785547 Free PMC article. Review.
-
Quantitative proteomic analysis of HER2 protein expression in PDAC tumors.Clin Proteomics. 2024 Mar 20;21(1):24. doi: 10.1186/s12014-024-09476-7. Clin Proteomics. 2024. PMID: 38509475 Free PMC article.
References
-
- Cuevas D, Velasco A, Vaquero M, Santacana M, Gatius S, Eritja N, et al. Intratumour heterogeneity in endometrial serous carcinoma assessed by targeted sequencing and multiplex ligation-dependent probe amplification: a descriptive study. Histopathology. 2020;76(3):447–460. doi: 10.1111/his.14001. - DOI - PubMed
Grants and funding
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
- HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027, and HU0001-22-2-0016/Defense Health Agency
LinkOut - more resources
Full Text Sources