

SURGE: uncovering context-specific genetic-regulation of gene expression from single-cell RNA sequencing using latent-factor models
- PMID: 38254214
- PMCID: PMC10801966
- DOI: 10.1186/s13059-023-03152-z
SURGE: uncovering context-specific genetic-regulation of gene expression from single-cell RNA sequencing using latent-factor models
Abstract
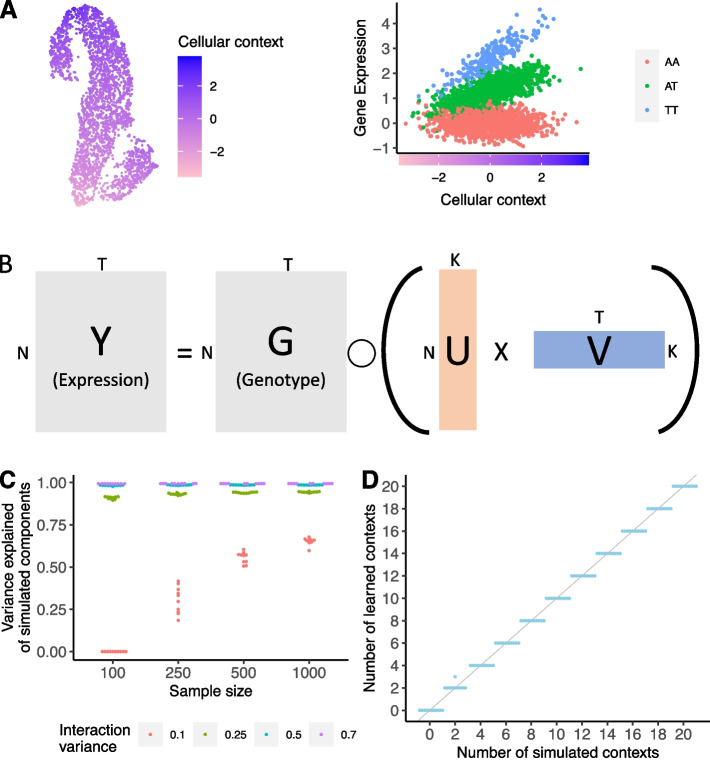
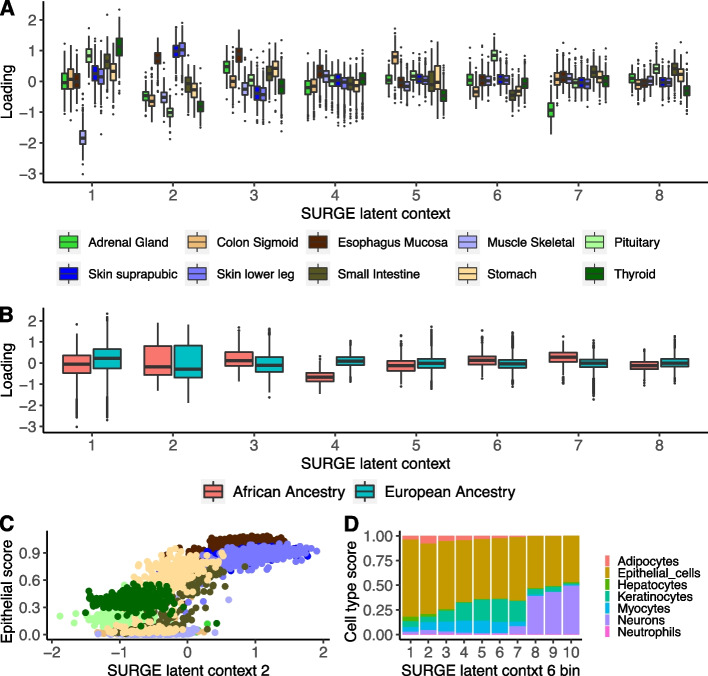
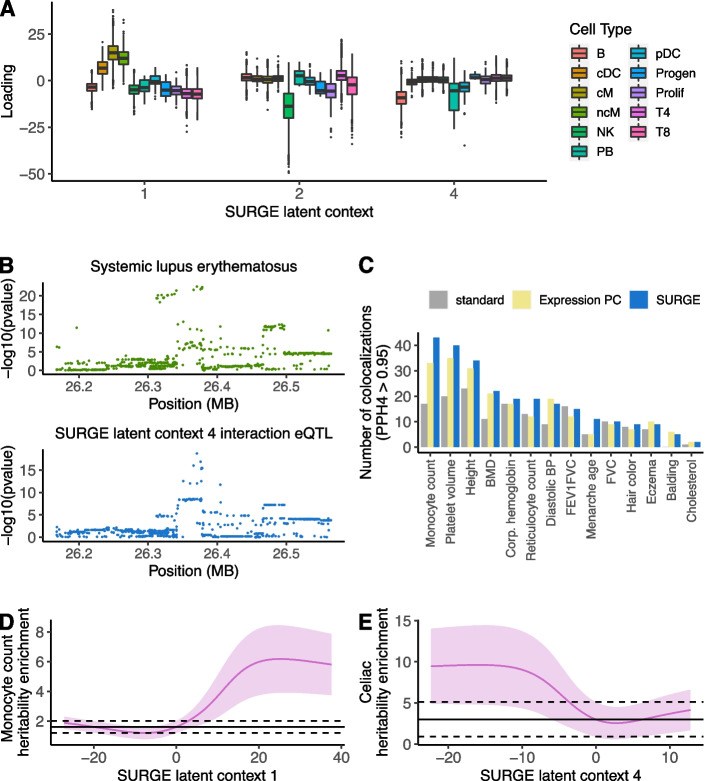
Genetic regulation of gene expression is a complex process, with genetic effects known to vary across cellular contexts such as cell types and environmental conditions. We developed SURGE, a method for unsupervised discovery of context-specific expression quantitative trait loci (eQTLs) from single-cell transcriptomic data. This allows discovery of the contexts or cell types modulating genetic regulation without prior knowledge. Applied to peripheral blood single-cell eQTL data, SURGE contexts capture continuous representations of distinct cell types and groupings of biologically related cell types. We demonstrate the disease-relevance of SURGE context-specific eQTLs using colocalization analysis and stratified LD-score regression.
Keywords: Single-cell transcriptomics; eQTL.
© 2024. The Author(s).
Conflict of interest statement
AB is a shareholder of Alphabet, Inc., and a consultant for Third Rock Ventures.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
