

Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson's Disease
- PMID: 38254673
- PMCID: PMC10813470
- DOI: 10.3390/biom14010073
Advancements in Genetic and Biochemical Insights: Unraveling the Etiopathogenesis of Neurodegeneration in Parkinson's Disease
Abstract
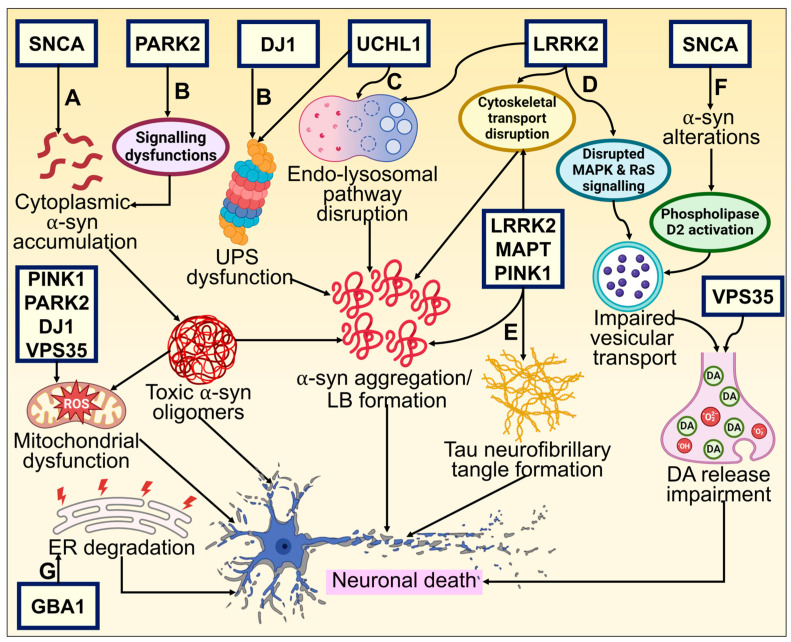
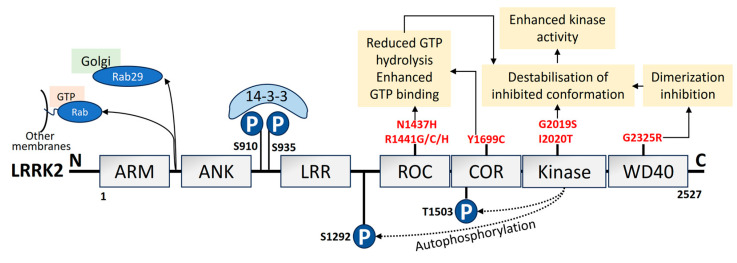
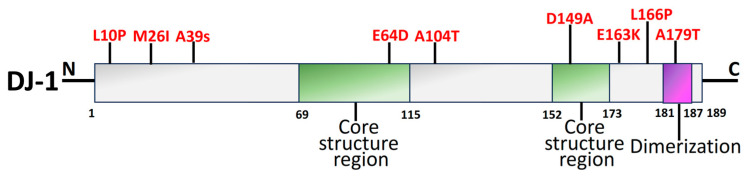
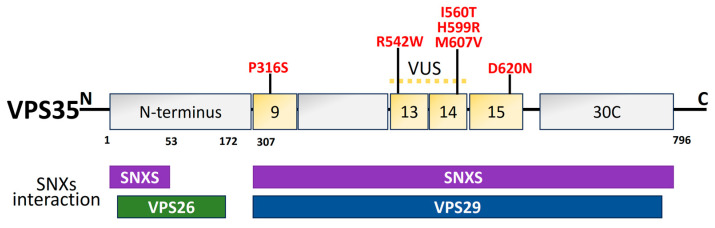
Parkinson's disease (PD) is the second most prevalent neurodegenerative movement disorder worldwide, which is primarily characterized by motor impairments. Even though multiple hypotheses have been proposed over the decades that explain the pathogenesis of PD, presently, there are no cures or promising preventive therapies for PD. This could be attributed to the intricate pathophysiology of PD and the poorly understood molecular mechanism. To address these challenges comprehensively, a thorough disease model is imperative for a nuanced understanding of PD's underlying pathogenic mechanisms. This review offers a detailed analysis of the current state of knowledge regarding the molecular mechanisms underlying the pathogenesis of PD, with a particular emphasis on the roles played by gene-based factors in the disease's development and progression. This study includes an extensive discussion of the proteins and mutations of primary genes that are linked to PD, including α-synuclein, GBA1, LRRK2, VPS35, PINK1, DJ-1, and Parkin. Further, this review explores plausible mechanisms for DAergic neural loss, non-motor and non-dopaminergic pathologies, and the risk factors associated with PD. The present study will encourage the related research fields to understand better and analyze the current status of the biochemical mechanisms of PD, which might contribute to the design and development of efficacious and safe treatment strategies for PD in future endeavors.
Keywords: Parkinson’s disease; genetics of Parkinson’s disease; molecular mechanism; risk factors; α-synuclein.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures






References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
