

The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy
- PMID: 38255995
- PMCID: PMC10815934
- DOI: 10.3390/ijms25020921
The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy
Abstract
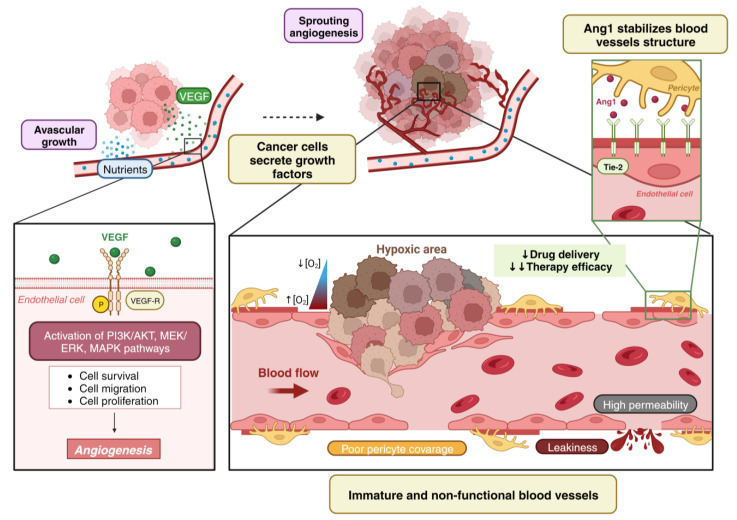
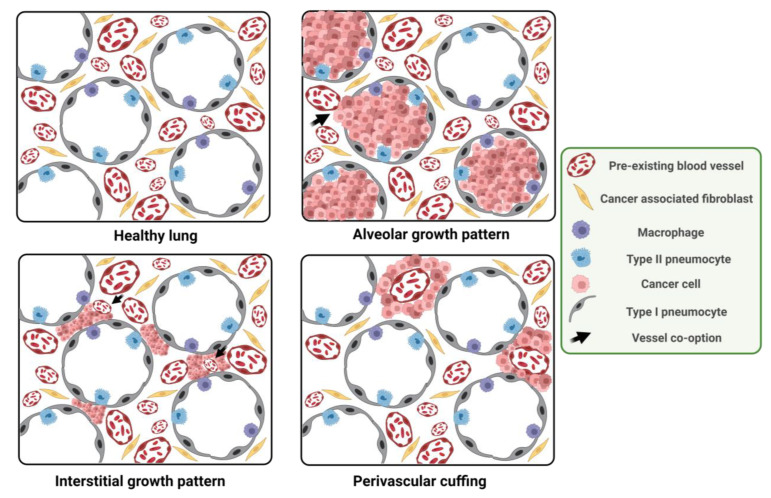
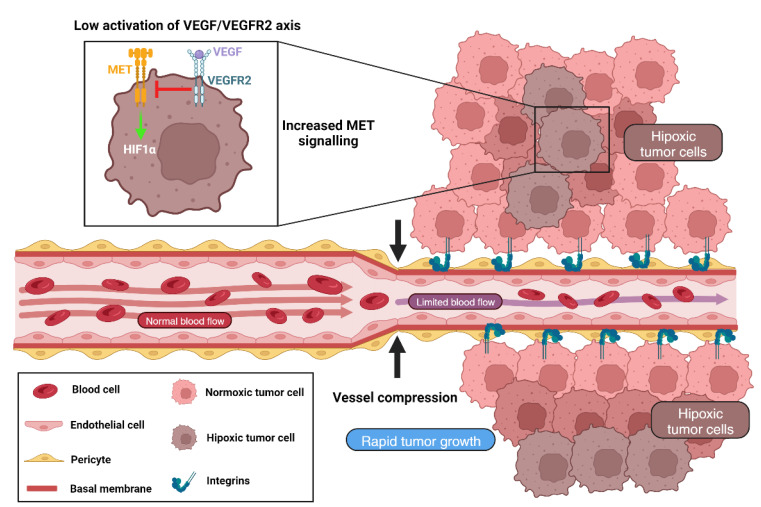
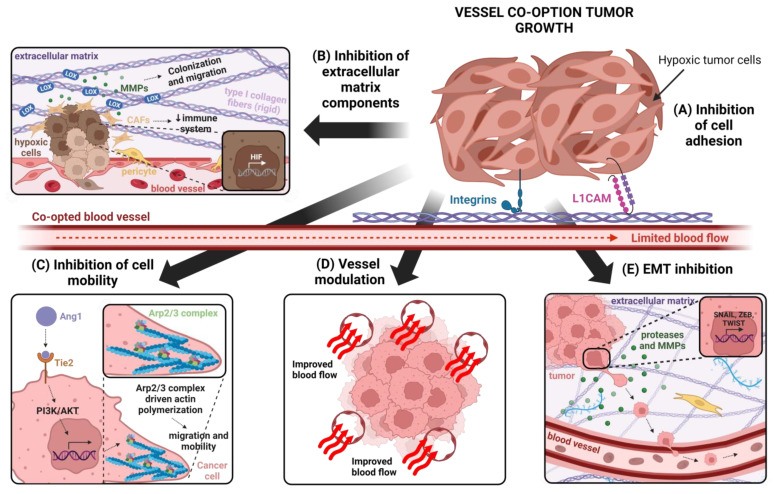
Vessel co-option (VCO) is a non-angiogenic mechanism of vascularization that has been associated to anti-angiogenic therapy. In VCO, cancer cells hijack the pre-existing blood vessels and use them to obtain oxygen and nutrients and invade adjacent tissue. Multiple primary tumors and metastases undergo VCO in highly vascularized tissues such as the lungs, liver or brain. VCO has been associated with a worse prognosis. The cellular and molecular mechanisms that undergo VCO are poorly understood. Recent studies have demonstrated that co-opted vessels show a quiescent phenotype in contrast to angiogenic tumor blood vessels. On the other hand, it is believed that during VCO, cancer cells are adhered to basement membrane from pre-existing blood vessels by using integrins, show enhanced motility and a mesenchymal phenotype. Other components of the tumor microenvironment (TME) such as extracellular matrix, immune cells or extracellular vesicles play important roles in vessel co-option maintenance. There are no strategies to inhibit VCO, and thus, to eliminate resistance to anti-angiogenic therapy. This review summarizes all the molecular mechanisms involved in vessel co-option analyzing the possible therapeutic strategies to inhibit this process.
Keywords: adhesion; angiogenesis; extracellular matrix; vessel co-option.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Tumor vessel co-option: The past & the future.Front Oncol. 2022 Aug 31;12:965277. doi: 10.3389/fonc.2022.965277. eCollection 2022. Front Oncol. 2022. PMID: 36119528 Free PMC article. Review.
-
Different Forms of Tumor Vascularization and Their Clinical Implications Focusing on Vessel Co-option in Colorectal Cancer Liver Metastases.Front Cell Dev Biol. 2021 Apr 12;9:612774. doi: 10.3389/fcell.2021.612774. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33912554 Free PMC article. Review.
-
Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models.J Pathol. 2017 Feb;241(3):362-374. doi: 10.1002/path.4845. Epub 2016 Dec 29. J Pathol. 2017. PMID: 27859259 Free PMC article.
-
Vessel co-option and resistance to anti-angiogenic therapy.Angiogenesis. 2020 Feb;23(1):55-74. doi: 10.1007/s10456-019-09698-6. Epub 2019 Dec 21. Angiogenesis. 2020. PMID: 31865479 Review.
-
Angiogenesis in NSCLC: is vessel co-option the trunk that sustains the branches?Oncotarget. 2017 Jun 13;8(24):39795-39804. doi: 10.18632/oncotarget.7794. Oncotarget. 2017. PMID: 26950275 Free PMC article. Review.
Cited by
-
Integrins as Key Mediators of Metastasis.Int J Mol Sci. 2025 Jan 22;26(3):904. doi: 10.3390/ijms26030904. Int J Mol Sci. 2025. PMID: 39940673 Free PMC article. Review.
-
Triple‑negative breast cancer cell‑derived piR‑31115 promotes the proliferation and migration of endothelial cells via METTL3‑mediated m6A modification of YAP1.Oncol Rep. 2025 Mar;53(3):34. doi: 10.3892/or.2025.8867. Epub 2025 Jan 17. Oncol Rep. 2025. PMID: 39820521 Free PMC article.
-
Glycosylation in cancer: mechanisms, diagnostic markers, and therapeutic applications.Mol Cell Biochem. 2025 May 19. doi: 10.1007/s11010-025-05303-1. Online ahead of print. Mol Cell Biochem. 2025. PMID: 40389792 Review.
-
Inhibition of Ovarian Cancer Growth, Metastasis and Reverse the Tumor Microenvironment by Dual Drug-Loaded Polymer Micelle Targeting Tumor Microenvironment.Int J Nanomedicine. 2025 Mar 12;20:2969-2990. doi: 10.2147/IJN.S507038. eCollection 2025. Int J Nanomedicine. 2025. PMID: 40098720 Free PMC article.
-
Role of Extracellular Vesicles in the Progression of Brain Tumors.Biology (Basel). 2024 Aug 2;13(8):586. doi: 10.3390/biology13080586. Biology (Basel). 2024. PMID: 39194524 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
