

The Role of SCAP/SREBP as Central Regulators of Lipid Metabolism in Hepatic Steatosis
- PMID: 38256181
- PMCID: PMC10815951
- DOI: 10.3390/ijms25021109
The Role of SCAP/SREBP as Central Regulators of Lipid Metabolism in Hepatic Steatosis
Abstract
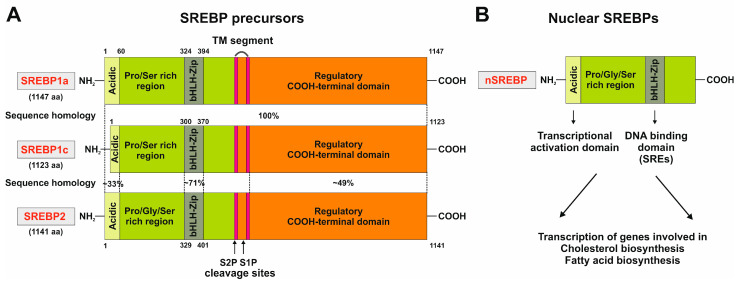
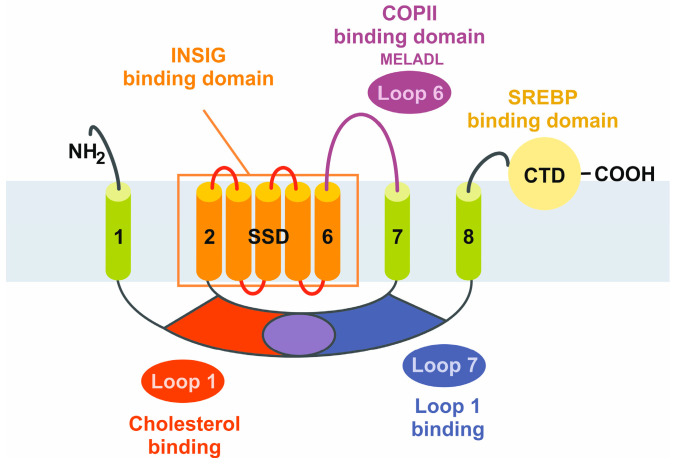
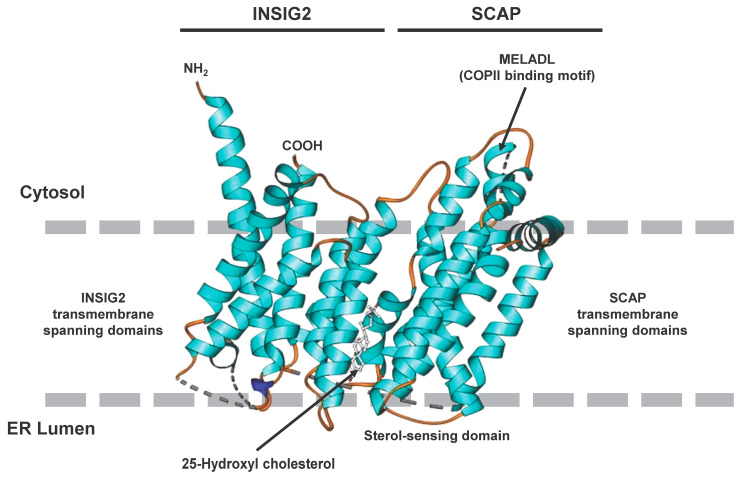
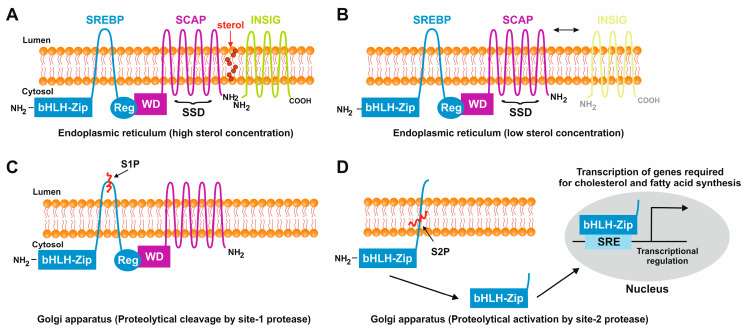
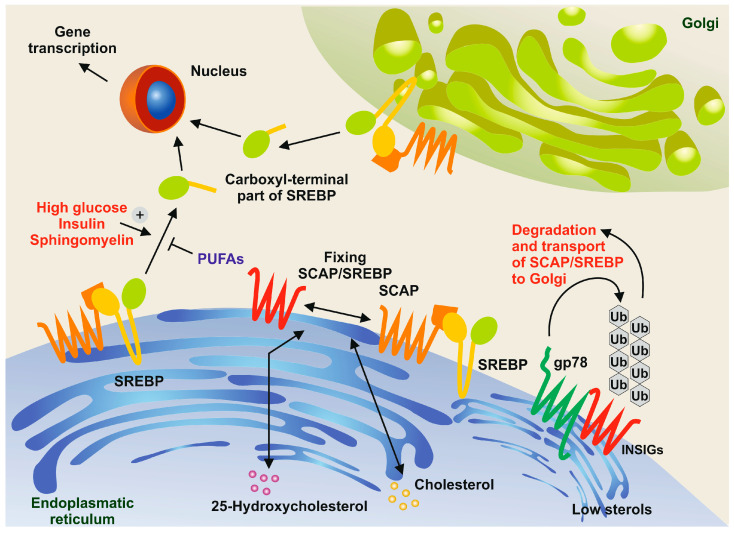
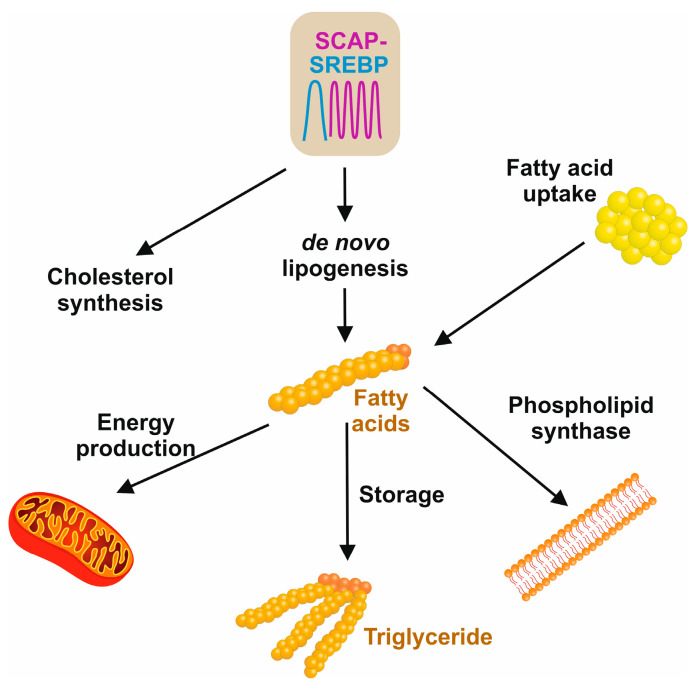
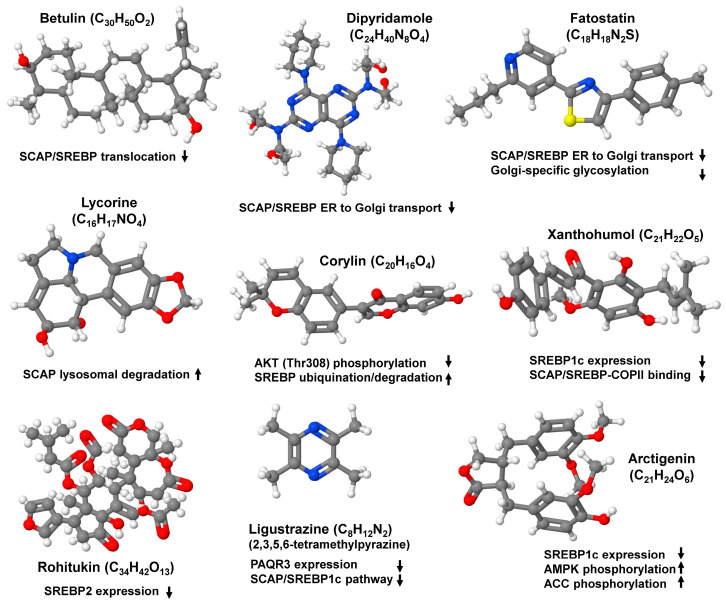
The prevalence of metabolic dysfunction-associated steatotic liver disease (MASLD) is rapidly increasing worldwide at an alarming pace, due to an increase in obesity, sedentary and unhealthy lifestyles, and unbalanced dietary habits. MASLD is a unique, multi-factorial condition with several phases of progression including steatosis, steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma. Sterol element binding protein 1c (SREBP1c) is the main transcription factor involved in regulating hepatic de novo lipogenesis. This transcription factor is synthesized as an inactive precursor, and its proteolytic maturation is initiated in the membrane of the endoplasmic reticulum upon stimulation by insulin. SREBP cleavage activating protein (SCAP) is required as a chaperon protein to escort SREBP from the endoplasmic reticulum and to facilitate the proteolytic release of the N-terminal domain of SREBP into the Golgi. SCAP inhibition prevents activation of SREBP and inhibits the expression of genes involved in triglyceride and fatty acid synthesis, resulting in the inhibition of de novo lipogenesis. In line, previous studies have shown that SCAP inhibition can resolve hepatic steatosis in animal models and intensive research is going on to understand the effects of SCAP in the pathogenesis of human disease. This review focuses on the versatile roles of SCAP/SREBP regulation in de novo lipogenesis and the structure and molecular features of SCAP/SREBP in the progression of hepatic steatosis. In addition, recent studies that attempt to target the SCAP/SREBP axis as a therapeutic option to interfere with MASLD are discussed.
Keywords: MASLD; SCAP; SREBP1c; de novo lipogenesis; fatty acid synthesis; insulin resistance; lipid metabolism; liver; pathogenesis; therapy; triglycerides.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Low abundance of insulin-induced gene 1 contributes to SREBP-1c processing and hepatic steatosis in dairy cows with severe fatty liver.J Dairy Sci. 2023 Aug;106(8):5626-5635. doi: 10.3168/jds.2022-22895. Epub 2023 Jun 7. J Dairy Sci. 2023. PMID: 37291038
-
Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c.Diabetes Obes Metab. 2010 Oct;12 Suppl 2:83-92. doi: 10.1111/j.1463-1326.2010.01275.x. Diabetes Obes Metab. 2010. PMID: 21029304 Review.
-
Understanding the Link Between Sterol Regulatory Element Binding Protein (SREBPs) and Metabolic Dysfunction Associated Steatotic Liver Disease (MASLD).Curr Obes Rep. 2025 Apr 14;14(1):36. doi: 10.1007/s13679-025-00626-y. Curr Obes Rep. 2025. PMID: 40227546 Review.
-
Lipogenesis and MASLD: re-thinking the role of SREBPs.Arch Toxicol. 2025 Jun;99(6):2299-2312. doi: 10.1007/s00204-025-04052-w. Epub 2025 May 6. Arch Toxicol. 2025. PMID: 40327083 Review.
-
Dipyridamole Inhibits Lipogenic Gene Expression by Retaining SCAP-SREBP in the Endoplasmic Reticulum.Cell Chem Biol. 2021 Feb 18;28(2):169-179.e7. doi: 10.1016/j.chembiol.2020.10.003. Epub 2020 Oct 22. Cell Chem Biol. 2021. PMID: 33096051 Free PMC article.
Cited by
-
Orchestration of Gut-Liver-Associated Transcription Factors in MAFLD: From Cross-Organ Interactions to Therapeutic Innovation.Biomedicines. 2025 Jun 10;13(6):1422. doi: 10.3390/biomedicines13061422. Biomedicines. 2025. PMID: 40564141 Free PMC article. Review.
-
Hepatic HSD17B6 is dispensable for diet-induced fatty liver disease in mice.Biochem Biophys Rep. 2025 Jan 19;41:101924. doi: 10.1016/j.bbrep.2025.101924. eCollection 2025 Mar. Biochem Biophys Rep. 2025. PMID: 39896111 Free PMC article.
-
Lipid metabolism: the potential therapeutic targets in glioblastoma.Cell Death Discov. 2025 Mar 17;11(1):107. doi: 10.1038/s41420-025-02390-3. Cell Death Discov. 2025. PMID: 40097417 Free PMC article. Review.
-
Cholesterol restriction primes antiviral innate immunity via SREBP1-driven noncanonical type I IFNs.EMBO Rep. 2025 Jan;26(2):560-592. doi: 10.1038/s44319-024-00346-9. Epub 2024 Dec 12. EMBO Rep. 2025. PMID: 39668245 Free PMC article.
-
The cellular and molecular targets of natural products against metabolic disorders: a translational approach to reach the bedside.MedComm (2020). 2024 Jul 24;5(8):e664. doi: 10.1002/mco2.664. eCollection 2024 Aug. MedComm (2020). 2024. PMID: 39049964 Free PMC article. Review.
References
-
- Ludwig J., Viggiano T.R., McGill D.B., Oh B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980;55:434–438. - PubMed
-
- Lovric A., Granér M., Bjornson E., Arif M., Benfeitas R., Nyman K., Ståhlman M., Pentikäinen M.O., Lundbom J., Hakkarainen A., et al. Characterization of different fat depots in NAFLD using inflammation-associated proteome, lipidome and metabolome. Sci. Rep. 2018;8:14200. doi: 10.1038/s41598-018-31865-w. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
