

Metagenomic Analysis of Viromes of Aedes Mosquitoes across India
- PMID: 38257809
- PMCID: PMC10818685
- DOI: 10.3390/v16010109
Metagenomic Analysis of Viromes of Aedes Mosquitoes across India
Abstract
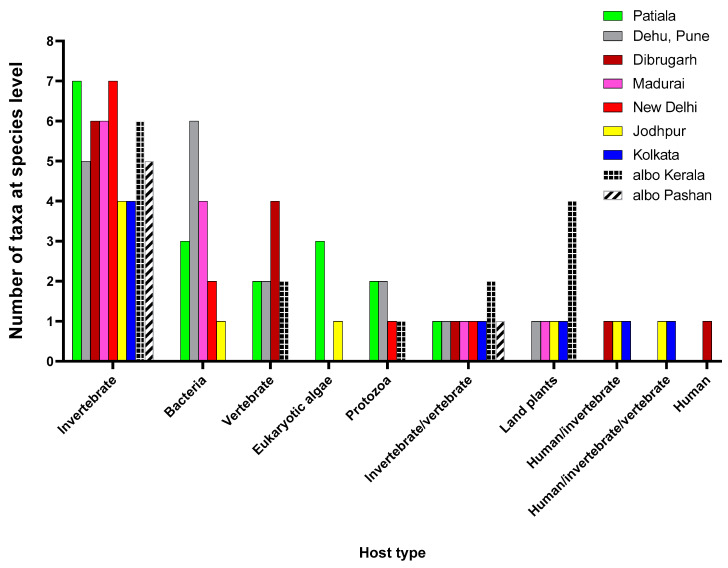
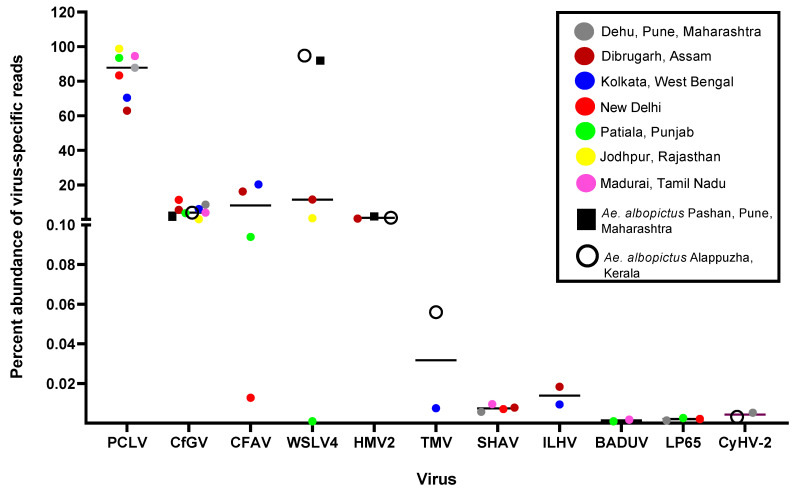
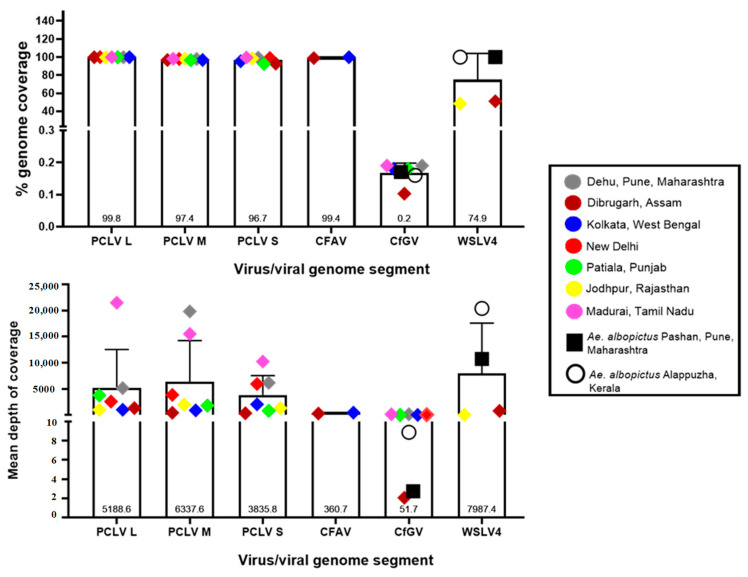
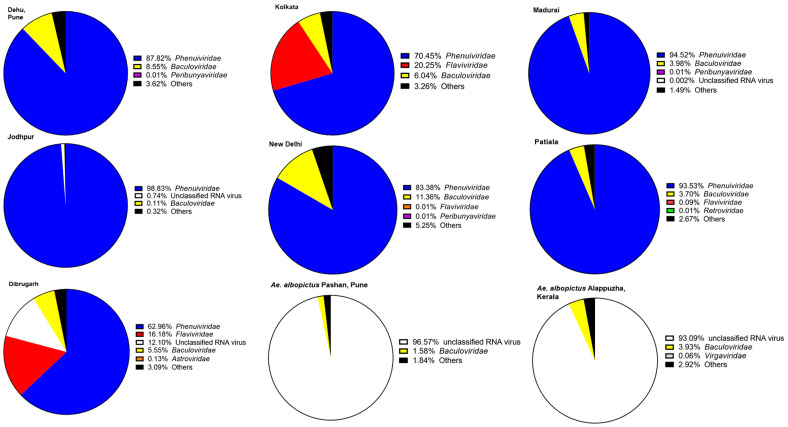
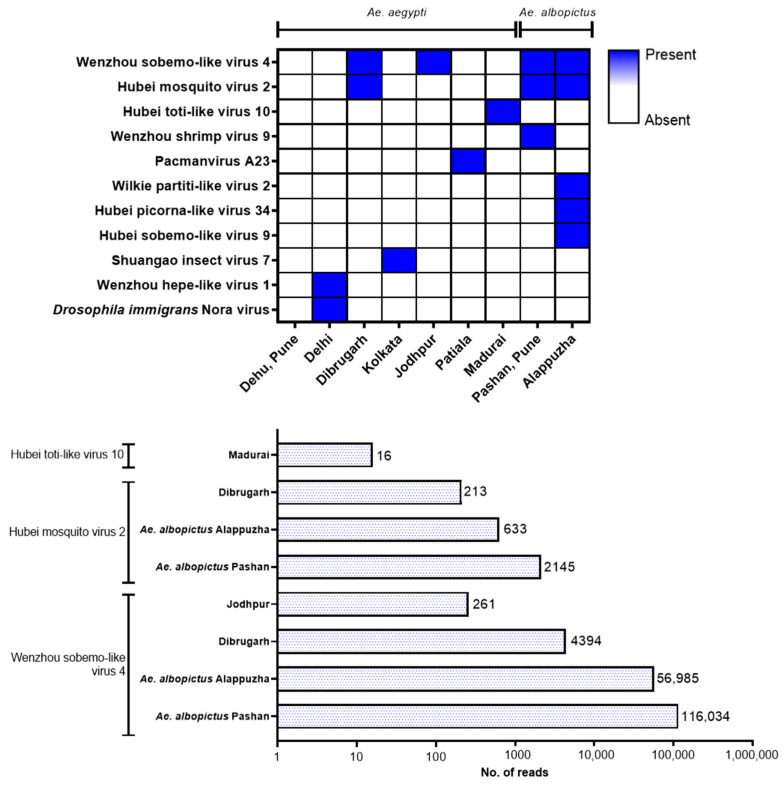
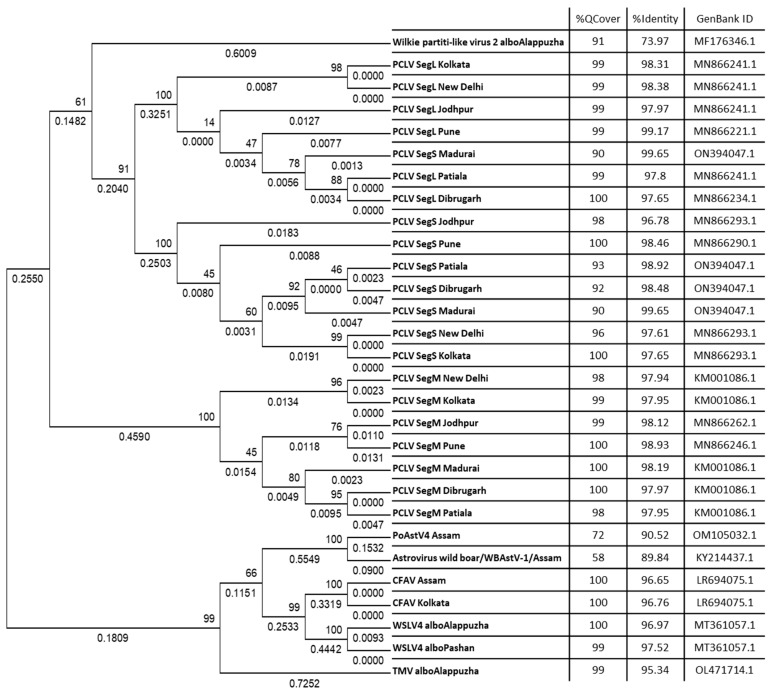
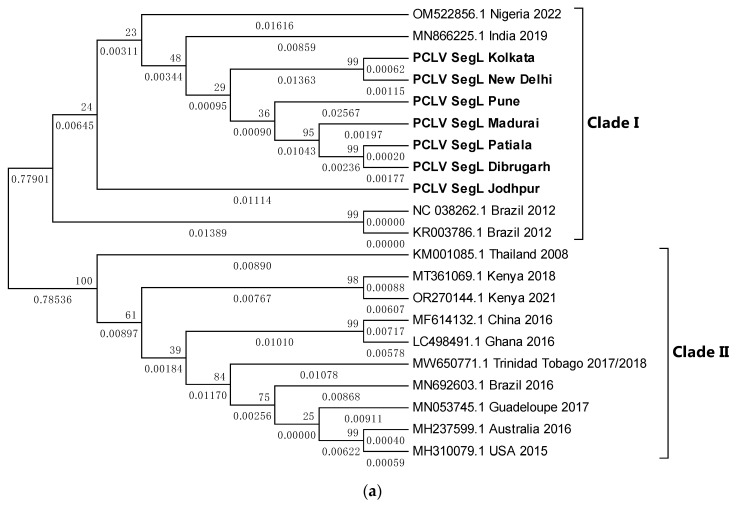
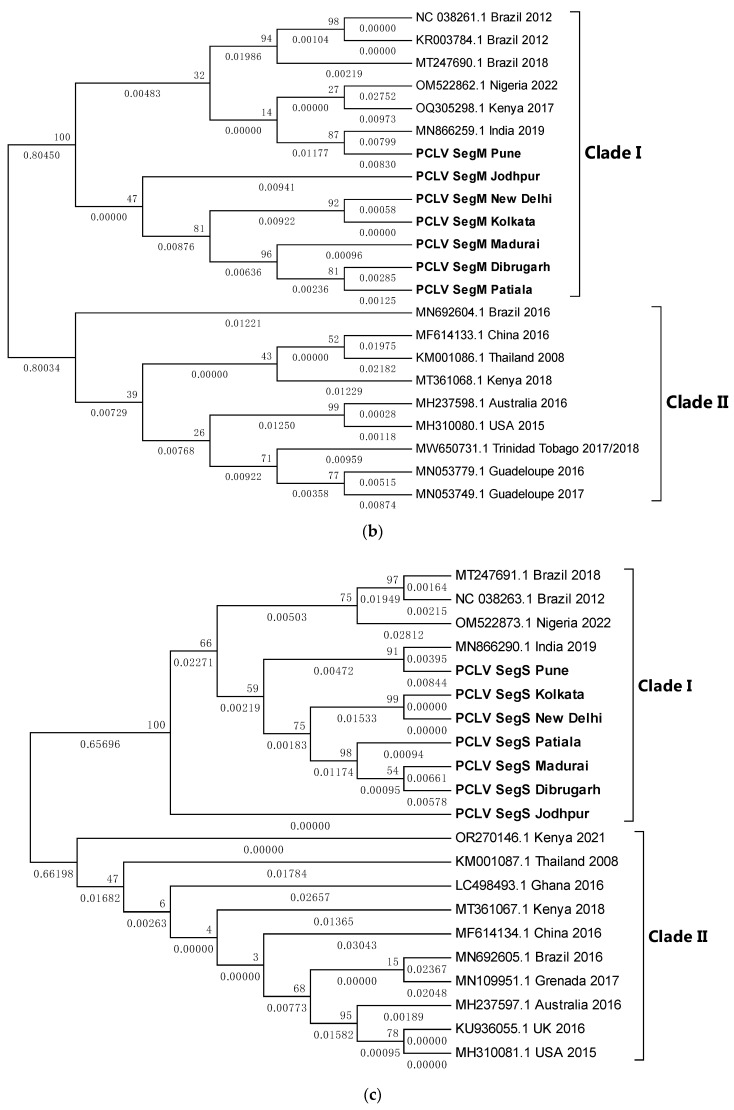
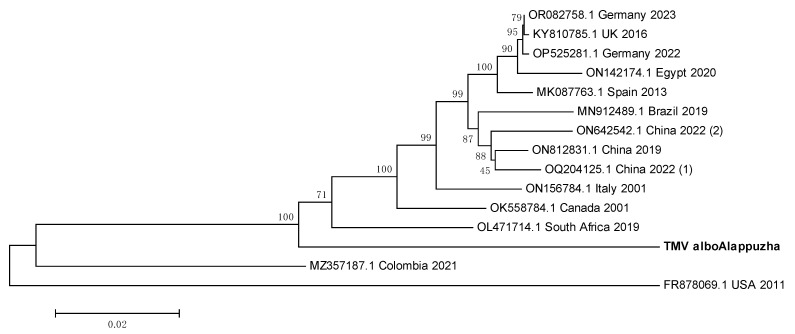
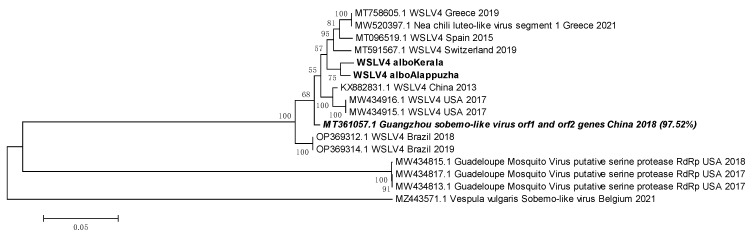
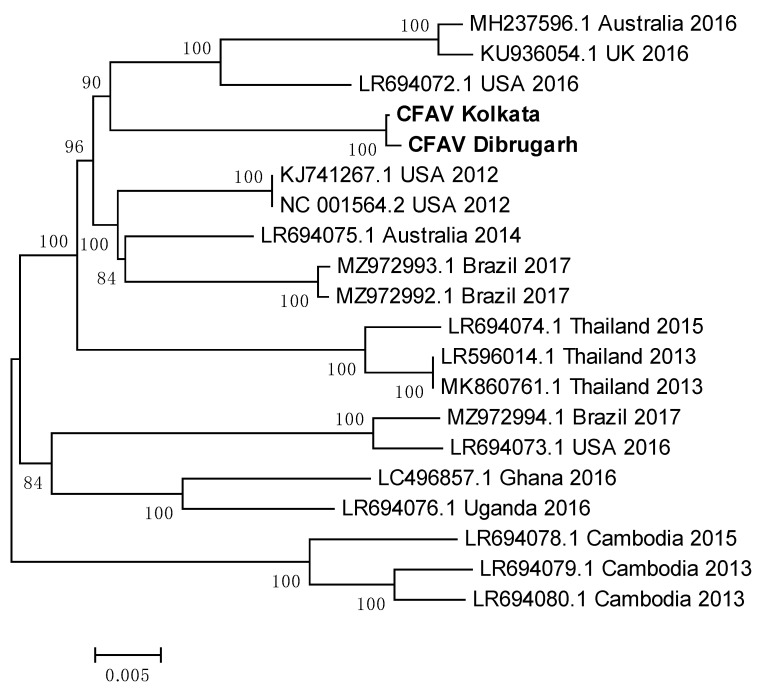
Metagenomic analysis of Aedes aegypti and Ae. albopictus mosquitoes from diverse geographical regions of India revealed the presence of several insect viruses of human interest. Most abundant reads found in Ae. aegypti mosquitoes were of Phasi Charoen-like virus (PCLV), Choristoneura fumiferana granulovirus (CfGV), Cell fusing agent virus (CFAV), and Wenzhou sobemo-like virus 4 (WSLV4), whereas WSLV4 and CfGV constituted the highest percentage of reads in Ae. albopictus viromes. Other reads that were of low percentage included Hubei mosquito virus 2 (HMV2), Porcine astrovirus 4 (PAstV4), and Wild Boar astrovirus (WBAstV). PCLV and CFAV, which were found to be abundant in Ae. aegypti viromes were absent in Ae. albopictus viromes. Among the viromes analyzed, Ae. aegypti sampled from Pune showed the highest percentage (79.82%) of viral reads, while Ae. aegypti mosquitoes sampled from Dibrugarh showed the lowest percentage (3.47%). Shamonda orthobunyavirus (SHAV), African swine fever virus (ASFV), Aroa virus (AROAV), and Ilheus virus (ILHV), having the potential to infect vertebrates, including humans, were also detected in both mosquito species, albeit with low read numbers. Reads of gemykibivirus, avian retrovirus, bacteriophages, herpesviruses, and viruses infecting protozoans, algae, etc., were also detected in the mosquitoes. A high percentage of reads in the Ae. albopictus mosquito samples belonged to unclassified viruses and warrant further investigation. The data generated in the present work may not only lead to studies to explain the influence of these viruses on the replication and transmission of viruses of clinical importance but also to find applications as biocontrol agents against pathogenic viruses.
Keywords: Aedes aegypti; Aedes albopictus; CFAV; CfGV; PCLV; Virome.
Conflict of interest statement
The authors have no relevant financial or non-financial interests to disclose.
Figures


Similar articles
-
Abundance of Phasi-Charoen-like virus in Aedes aegypti mosquito populations in different states of India.PLoS One. 2022 Dec 9;17(12):e0277276. doi: 10.1371/journal.pone.0277276. eCollection 2022. PLoS One. 2022. PMID: 36490242 Free PMC article.
-
Local-scale virome depiction in Medellín, Colombia, supports significant differences between Aedes aegypti and Aedes albopictus.PLoS One. 2022 Jul 27;17(7):e0263143. doi: 10.1371/journal.pone.0263143. eCollection 2022. PLoS One. 2022. PMID: 35895627 Free PMC article.
-
Influence of dengue virus serotypes on the abundance of Aedes aegypti insect-specific viruses (ISVs).J Virol. 2024 Jan 23;98(1):e0150723. doi: 10.1128/jvi.01507-23. Epub 2023 Dec 14. J Virol. 2024. PMID: 38095414 Free PMC article.
-
Stability of the Virome in Lab- and Field-Collected Aedes albopictus Mosquitoes across Different Developmental Stages and Possible Core Viruses in the Publicly Available Virome Data of Aedes Mosquitoes.mSystems. 2020 Sep 29;5(5):e00640-20. doi: 10.1128/mSystems.00640-20. mSystems. 2020. PMID: 32994288 Free PMC article.
-
Aedes aegypti and Ae. albopictus microbiome/virome: new strategies for controlling arboviral transmission?Parasit Vectors. 2022 Aug 9;15(1):287. doi: 10.1186/s13071-022-05401-9. Parasit Vectors. 2022. PMID: 35945559 Free PMC article. Review.
Cited by
-
Investigation of RNA Viruses in Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) in a Mining Complex in the Southeastern Region of the Brazilian Amazon.Viruses. 2024 Nov 29;16(12):1862. doi: 10.3390/v16121862. Viruses. 2024. PMID: 39772171 Free PMC article.
-
Characterization of the Virome in Mosquitoes Across Distinct Habitats in the Yucatán Peninsula, Mexico.Viruses. 2025 May 26;17(6):758. doi: 10.3390/v17060758. Viruses. 2025. PMID: 40573349 Free PMC article.
-
Aedes Mosquito Virome in Southwestern Cameroon: Lack of Core Virome, But a Very Rich and Diverse Virome in Ae. africanus Compared to Other Aedes Species.Viruses. 2024 Jul 21;16(7):1172. doi: 10.3390/v16071172. Viruses. 2024. PMID: 39066334 Free PMC article.
-
First isolation and full-length genome analysis of a new RNA virus, Wenzhou sobemo-like virus 4 from Culex tritaeniorhynchus.Virulence. 2025 Dec;16(1):2539210. doi: 10.1080/21505594.2025.2539210. Epub 2025 Aug 4. Virulence. 2025. PMID: 40755044 Free PMC article.
-
Mapping Viral Landscapes: Genomic Surveillance of Aedes Mosquitoes From Central India.Cureus. 2025 Feb 18;17(2):e79206. doi: 10.7759/cureus.79206. eCollection 2025 Feb. Cureus. 2025. PMID: 40115680 Free PMC article.
References
-
- Fredericks A.C., Russell T.A., Wallace L.E., Davidson A.D., Fernandez-Sesma A., Maringer K. Aedes aegypti (Aag2)-Derived Clonal Mosquito Cell Lines Reveal the Effects of Pre-Existing Persistent Infection with the Insect-Specific Bunyavirus Phasi Charoen-like Virus on Arbovirus Replication. PLoS Negl. Trop. Dis. 2019;13:e0007346. doi: 10.1371/journal.pntd.0007346. - DOI - PMC - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
