

Discovery of novel and potent CDK8 inhibitors for the treatment of acute myeloid leukaemia
- PMID: 38258519
- PMCID: PMC10810651
- DOI: 10.1080/14756366.2024.2305852
Discovery of novel and potent CDK8 inhibitors for the treatment of acute myeloid leukaemia
Abstract
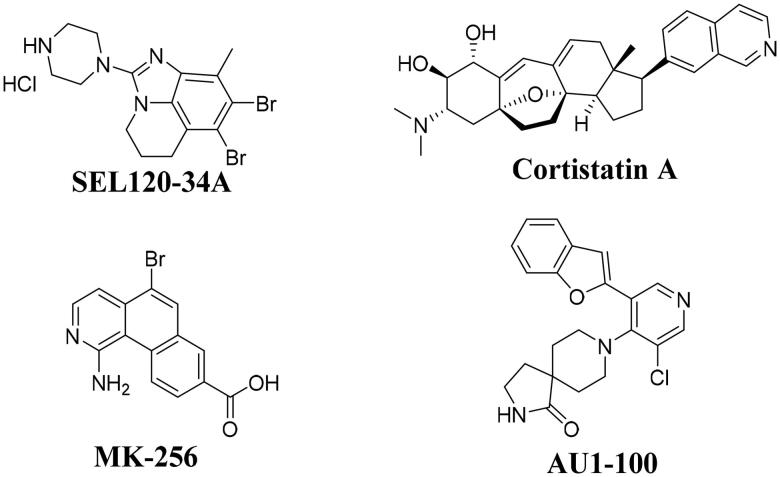
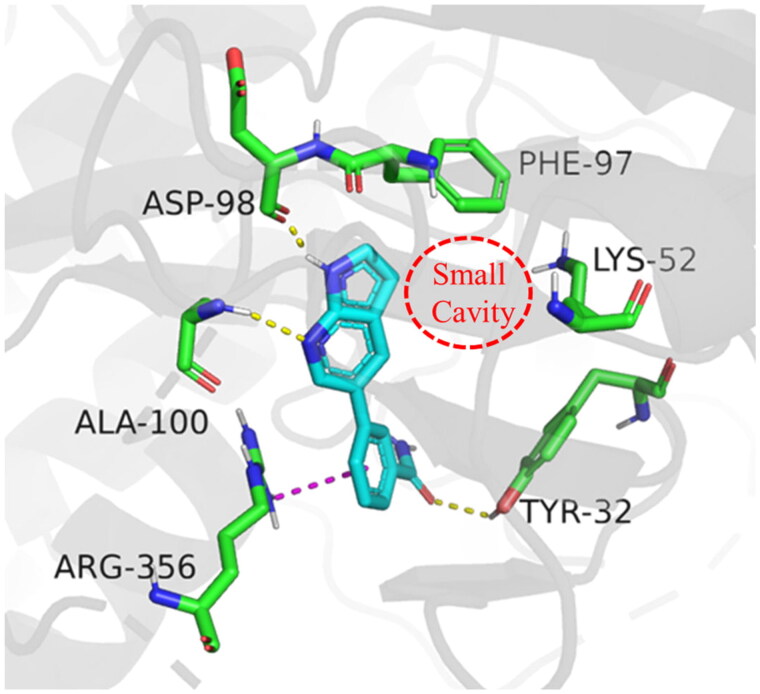
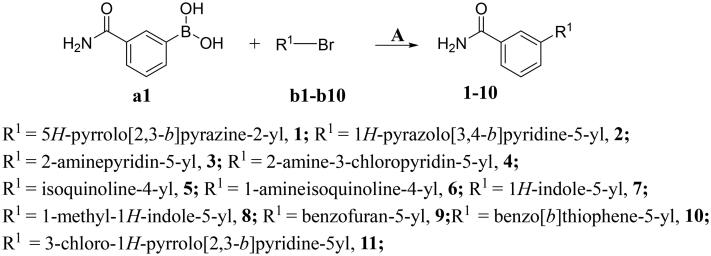
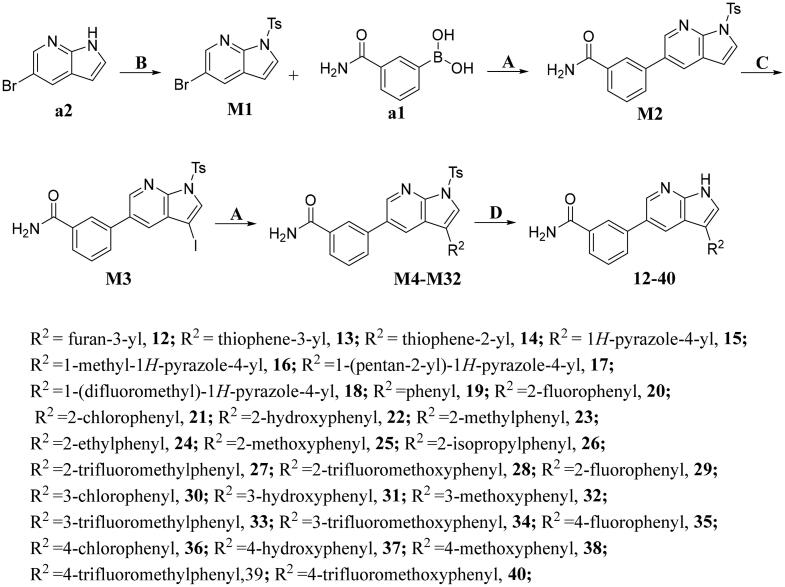
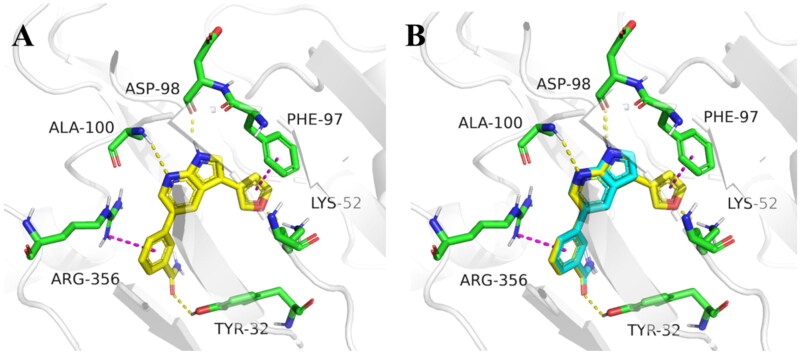
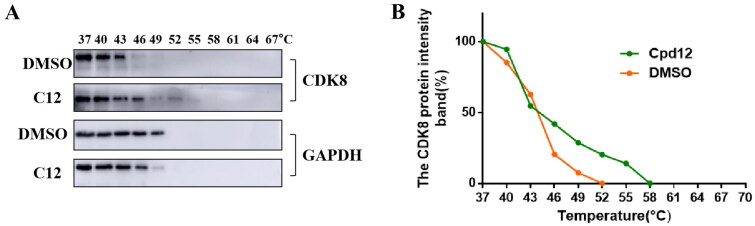
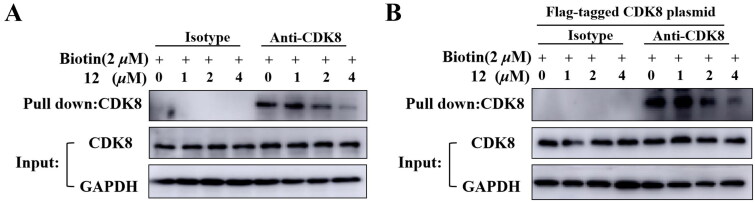
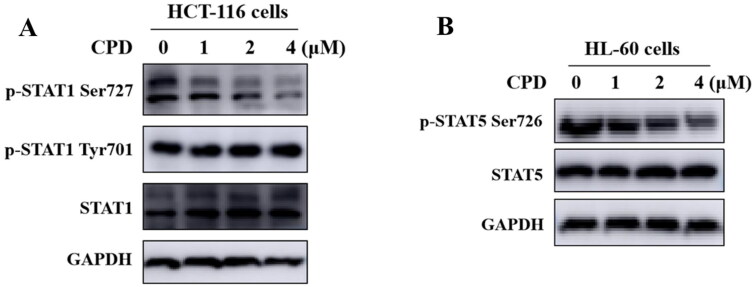
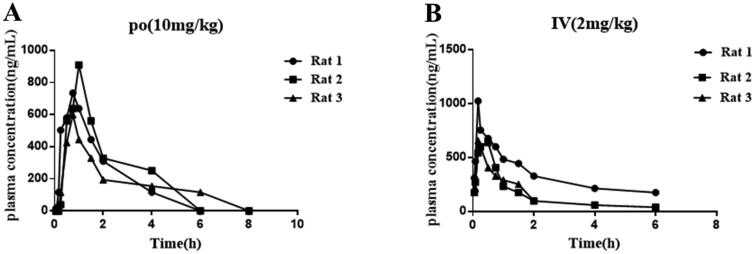
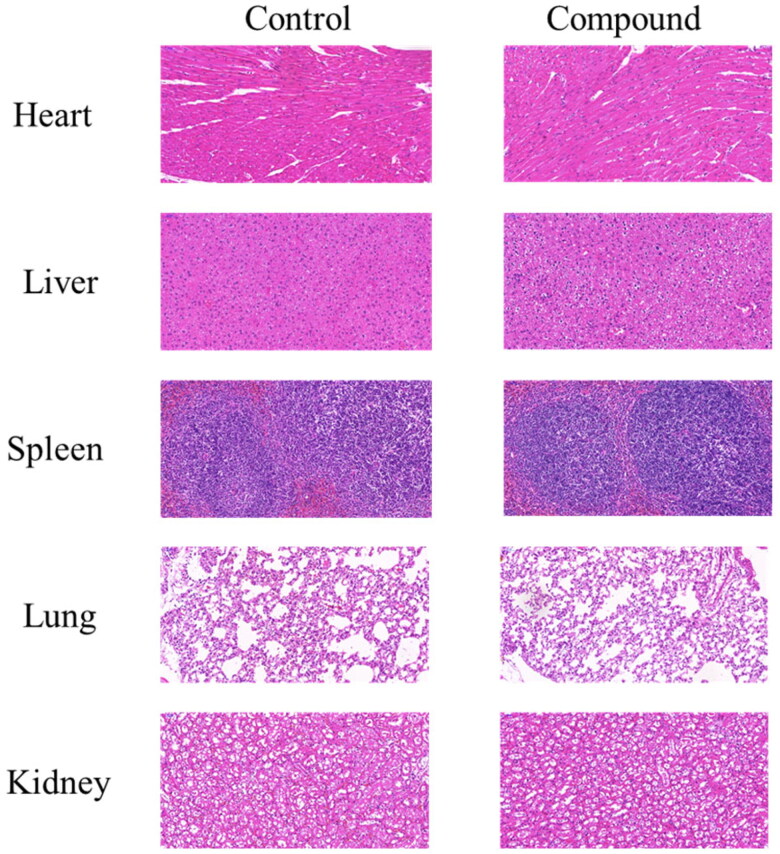
It has been reported that CDK8 plays a key role in acute myeloid leukaemia. Here, a total of 40 compounds were rational designed and synthesised based on the previous SAR. Among them, compound 12 (3-(3-(furan-3-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzamide) showed the most potent inhibiting activity against CDK8 with an IC50 value of 39.2 ± 6.3 nM and anti AML cell proliferation activity (molm-13 GC50 = 0.02 ± 0.01 μM, MV4-11 GC50 = 0.03 ± 0.01 μM). Mechanistic studies revealed that this compound 12 could inhibit the phosphorylation of STAT-1 and STAT-5. Importantly, compound 12 showed relative good bioavailability (F = 38.80%) and low toxicity in vivo. This study has great significance for the discovery of more efficient CDK8 inhibitors and the development of drugs for treating AML in the future.
Keywords: AML; CDK8 inhibitor; STAT-1; STAT-5.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
Similar articles
-
Design and synthesis of 7-azaindole derivatives as potent CDK8 inhibitors for the treatment of acute myeloid leukemia.RSC Med Chem. 2024 Jul 17;15(9):3180-95. doi: 10.1039/d4md00465e. Online ahead of print. RSC Med Chem. 2024. PMID: 39157854 Free PMC article.
-
Discovery of a novel oral type Ⅰ CDK8 inhibitor against acute myeloid leukemia.Eur J Med Chem. 2023 May 5;251:115214. doi: 10.1016/j.ejmech.2023.115214. Epub 2023 Feb 24. Eur J Med Chem. 2023. PMID: 36889252
-
MK256 is a novel CDK8 inhibitor with potent antitumor activity in AML through downregulation of the STAT pathway.Oncotarget. 2022 Nov 2;13:1217-1236. doi: 10.18632/oncotarget.28305. Oncotarget. 2022. PMID: 36342456 Free PMC article.
-
Discovery and Development of Cyclin-Dependent Kinase 8 Inhibitors.Curr Med Chem. 2020;27(32):5429-5443. doi: 10.2174/0929867326666190402110528. Curr Med Chem. 2020. PMID: 30947649 Review.
-
Binding patterns and structure-activity relationship of CDK8 inhibitors.Bioorg Chem. 2020 Mar;96:103624. doi: 10.1016/j.bioorg.2020.103624. Epub 2020 Jan 25. Bioorg Chem. 2020. PMID: 32078847 Review.
Cited by
-
Design and synthesis of 7-azaindole derivatives as potent CDK8 inhibitors for the treatment of acute myeloid leukemia.RSC Med Chem. 2024 Jul 17;15(9):3180-95. doi: 10.1039/d4md00465e. Online ahead of print. RSC Med Chem. 2024. PMID: 39157854 Free PMC article.
-
Cyclin-Dependent Kinase 8 Represents a Positive Regulator of Cytomegalovirus Replication and a Novel Host Target for Antiviral Strategies.Pharmaceutics. 2024 Sep 23;16(9):1238. doi: 10.3390/pharmaceutics16091238. Pharmaceutics. 2024. PMID: 39339274 Free PMC article.
-
Lysosome-derived biomarkers for predicting survival outcome in acute myeloid leukemia.Discov Oncol. 2025 Aug 2;16(1):1455. doi: 10.1007/s12672-025-03302-8. Discov Oncol. 2025. PMID: 40751887 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous