

Targeted genomic sequencing of avian influenza viruses in wetland sediment from wild bird habitats
- PMID: 38259077
- PMCID: PMC10880596
- DOI: 10.1128/aem.00842-23
Targeted genomic sequencing of avian influenza viruses in wetland sediment from wild bird habitats
Abstract
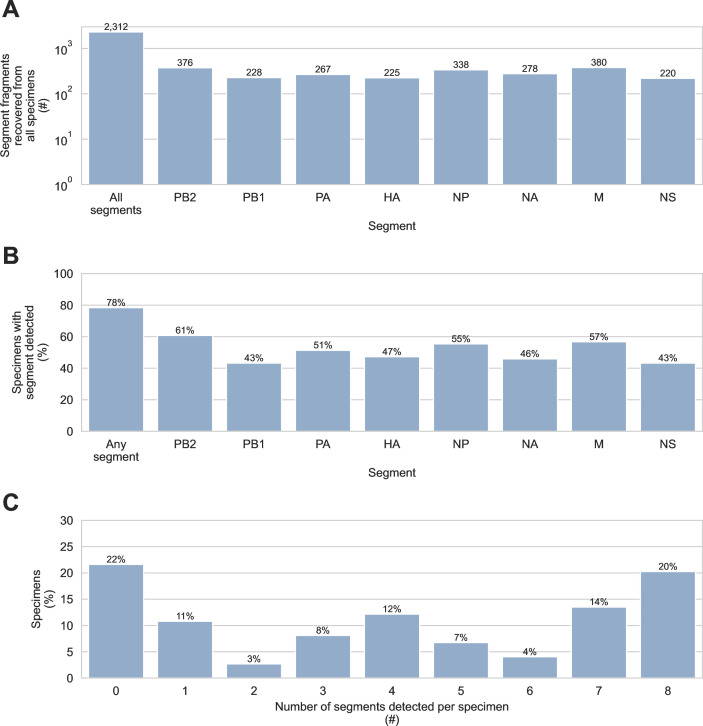
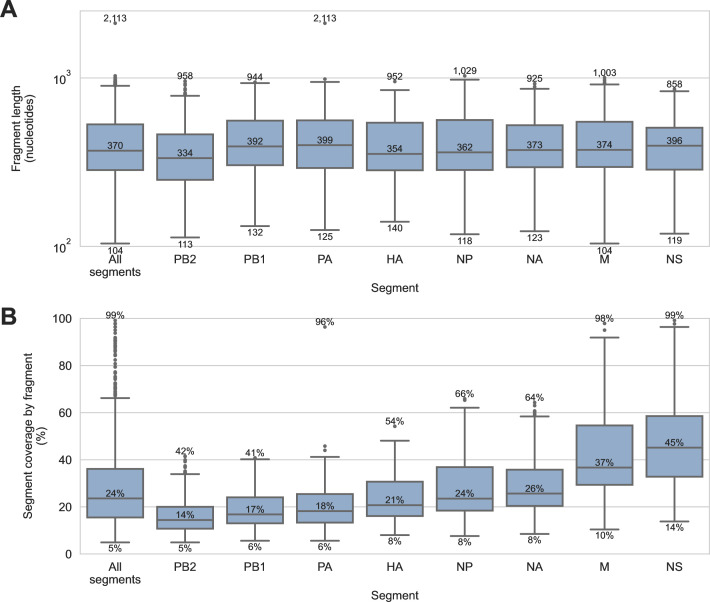
Diverse influenza A viruses (IAVs) circulate in wild birds, including highly pathogenic strains that infect poultry and humans. Consequently, surveillance of IAVs in wild birds is a cornerstone of agricultural biosecurity and pandemic preparedness. Surveillance is traditionally done by testing wild birds directly, but obtaining these specimens is labor intensive, detection rates can be low, and sampling is often biased toward certain avian species. As a result, local incursions of dangerous IAVs are rarely detected before outbreaks begin. Testing environmental specimens from wild bird habitats has been proposed as an alternative surveillance strategy. These specimens are thought to contain diverse IAVs deposited by a broad range of avian hosts, including species that are not typically sampled by surveillance programs. To enable this surveillance strategy, we developed a targeted genomic sequencing method for characterizing IAVs in these challenging environmental specimens. It combines custom hybridization probes, unique molecular index-based library construction, and purpose-built bioinformatic tools, allowing IAV genomic material to be enriched and analyzed with single-fragment resolution. We demonstrated our method on 90 sediment specimens from wetlands around Vancouver, Canada. We recovered 2,312 IAV genome fragments originating from all eight IAV genome segments. Eleven hemagglutinin subtypes and nine neuraminidase subtypes were detected, including H5, the current global surveillance priority. Our results demonstrate that targeted genomic sequencing of environmental specimens from wild bird habitats could become a valuable complement to avian influenza surveillance programs.IMPORTANCEIn this study, we developed genome sequencing tools for characterizing avian influenza viruses in sediment from wild bird habitats. These tools enable an environment-based approach to avian influenza surveillance. This could improve early detection of dangerous strains in local wild birds, allowing poultry producers to better protect their flocks and prevent human exposures to potential pandemic threats. Furthermore, we purposefully developed these methods to contend with viral genomic material that is diluted, fragmented, incomplete, and derived from multiple strains and hosts. These challenges are common to many environmental specimens, making these methods broadly applicable for genomic pathogen surveillance in diverse contexts.
Keywords: avian influenza; genomics; influenza; surveillance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Genetic Evidence Supports Sporadic and Independent Introductions of Subtype H5 Low-Pathogenic Avian Influenza A Viruses from Wild Birds to Domestic Poultry in North America.J Virol. 2018 Sep 12;92(19):e00913-18. doi: 10.1128/JVI.00913-18. Print 2018 Oct 1. J Virol. 2018. PMID: 30045988 Free PMC article.
-
Low-Pathogenic Influenza A Viruses in North American Diving Ducks Contribute to the Emergence of a Novel Highly Pathogenic Influenza A(H7N8) Virus.J Virol. 2017 Apr 13;91(9):e02208-16. doi: 10.1128/JVI.02208-16. Print 2017 May 1. J Virol. 2017. PMID: 28202755 Free PMC article.
-
Aerosol Transmission of Gull-Origin Iceland Subtype H10N7 Influenza A Virus in Ferrets.J Virol. 2019 Jun 14;93(13):e00282-19. doi: 10.1128/JVI.00282-19. Print 2019 Jul 1. J Virol. 2019. PMID: 30996092 Free PMC article.
-
Ecology of Influenza A Viruses in Wild Birds and Wetlands of Alaska.Avian Dis. 2020 Jun;64(2):109-122. doi: 10.1637/0005-2086-64.2.109. Avian Dis. 2020. PMID: 32550610 Review.
-
A Review of Avian Influenza A Virus Associations in Synanthropic Birds.Viruses. 2020 Oct 23;12(11):1209. doi: 10.3390/v12111209. Viruses. 2020. PMID: 33114239 Free PMC article. Review.
Cited by
-
Improved influenza A whole-genome sequencing protocol.Front Cell Infect Microbiol. 2024 Nov 28;14:1497278. doi: 10.3389/fcimb.2024.1497278. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 39669272 Free PMC article.
-
The role of artificial intelligence in detecting avian influenza virus outbreaks: A review.Open Vet J. 2025 May;15(5):1880-1894. doi: 10.5455/OVJ.2025.v15.i5.4. Epub 2025 May 31. Open Vet J. 2025. PMID: 40557099 Free PMC article. Review.
-
Transmission dynamics of highly pathogenic avian influenza virus at the wildlife-poultry-environmental interface: A case study.One Health. 2024 Nov 12;19:100932. doi: 10.1016/j.onehlt.2024.100932. eCollection 2024 Dec. One Health. 2024. PMID: 39640906 Free PMC article.
-
Descriptive Epidemiology and Phylodynamics of the "First Wave" of an Outbreak of Highly Pathogenic Avian Influenza (H5N1 Clade 2.3.4.4b) in British Columbia and the Yukon, Canada, April to September 2022.Transbound Emerg Dis. 2024 Feb 29;2024:2327939. doi: 10.1155/2024/2327939. eCollection 2024. Transbound Emerg Dis. 2024. PMID: 40303032 Free PMC article.
-
Descriptive epidemiology and phylogenetic analysis of highly pathogenic avian influenza H5N1 clade 2.3.4.4b in British Columbia (B.C.) and the Yukon, Canada, September 2022 to June 2023.Emerg Microbes Infect. 2024 Dec;13(1):2392667. doi: 10.1080/22221751.2024.2392667. Epub 2024 Sep 22. Emerg Microbes Infect. 2024. PMID: 39143912 Free PMC article.
References
-
- Ramos S. 2017. Impacts of the 2014-2015 highly pathogenic avian influenza outbreak on the U.S. poultry sector. LDPM-282-02:22. United States Department of Agriculture, Economic Research Service.
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
