

Decoding the secrets: how conformational and structural regulators inhibit the human 20S proteasome
- PMID: 38260042
- PMCID: PMC10801056
- DOI: 10.3389/fchem.2023.1322628
Decoding the secrets: how conformational and structural regulators inhibit the human 20S proteasome
Abstract
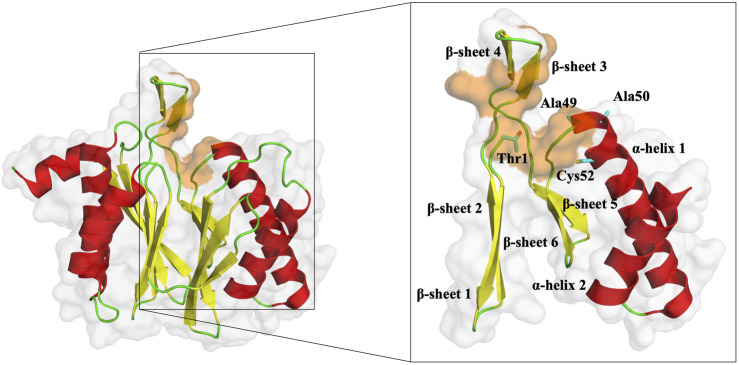
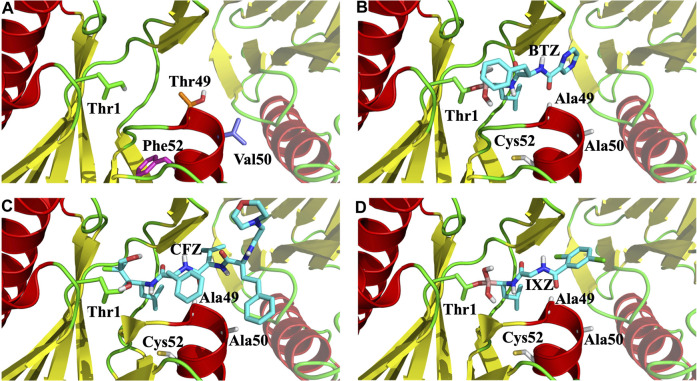
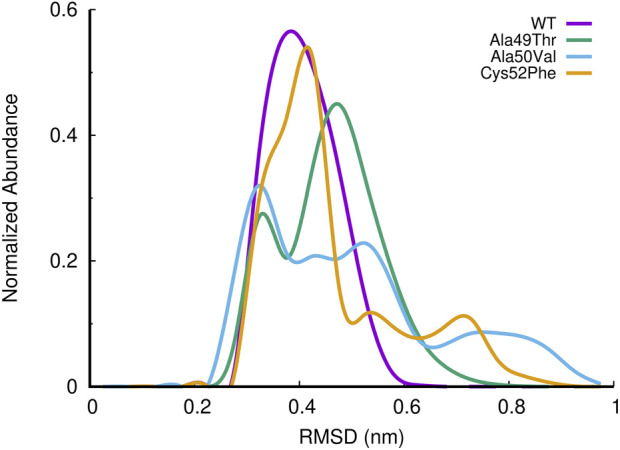
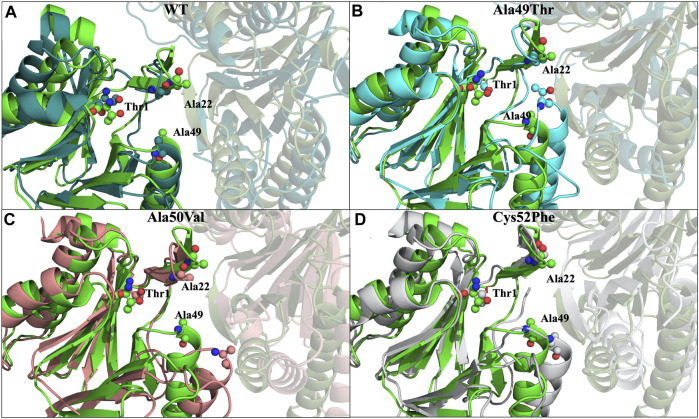
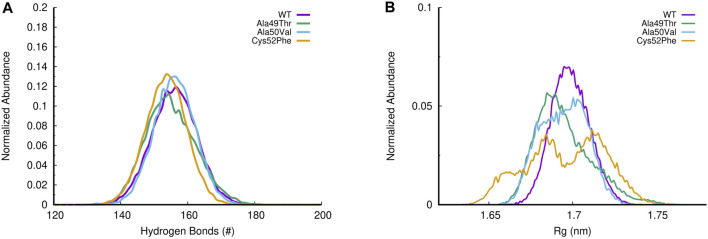
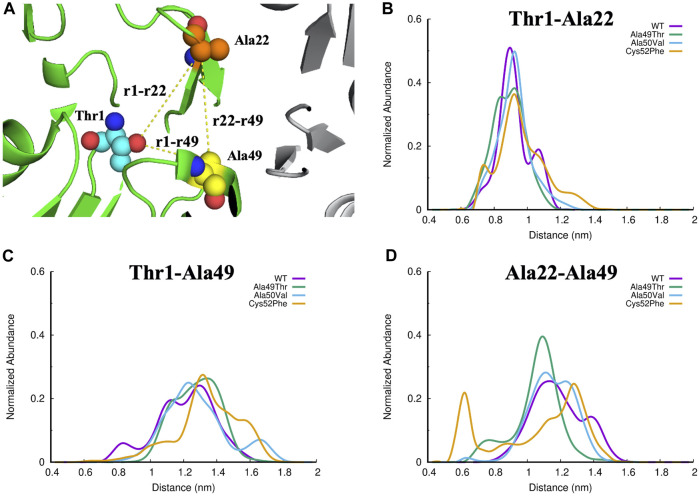
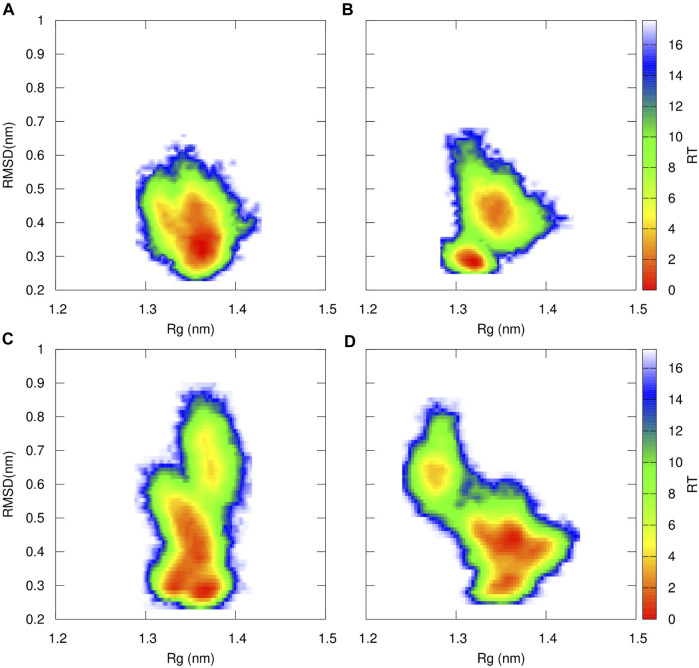
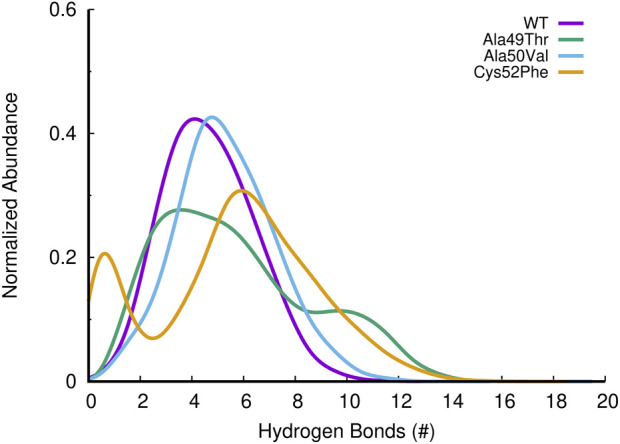
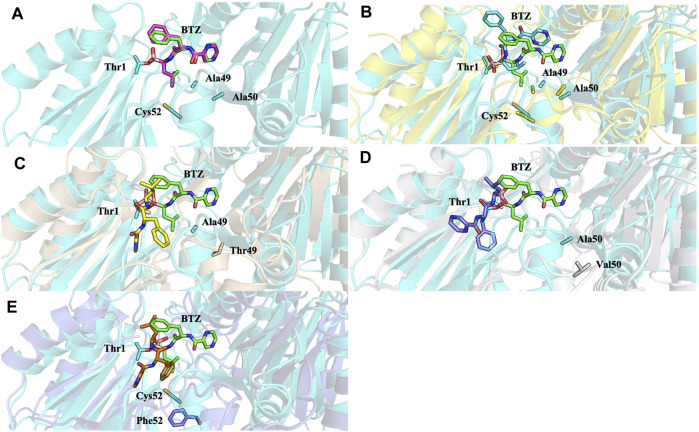
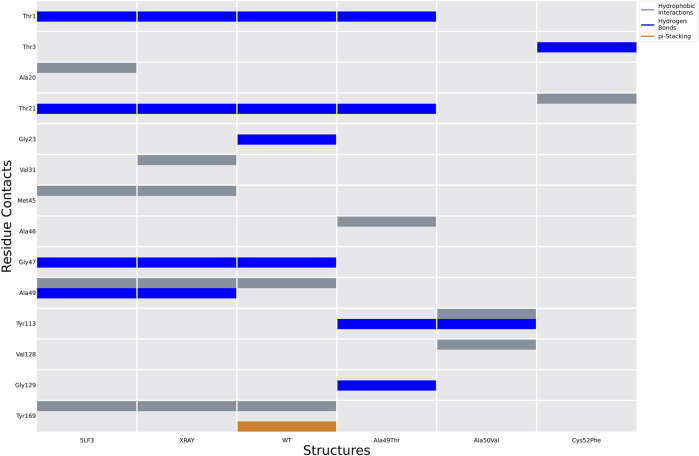
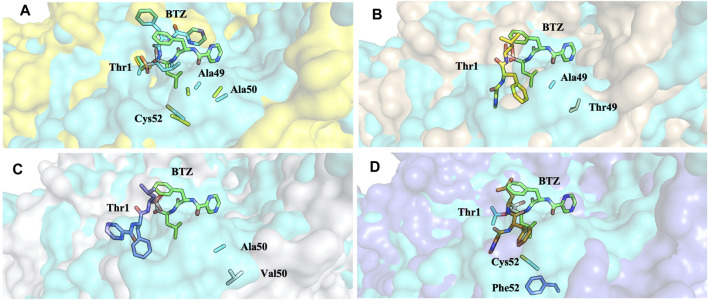
Acquired resistance to drugs that modulate specific protein functions, such as the human proteasome, presents a significant challenge in targeted therapies. This underscores the importance of devising new methodologies to predict drug binding and potential resistance due to specific protein mutations. In this work, we conducted an extensive computational analysis to ascertain the effects of selected mutations (Ala49Thr, Ala50Val, and Cys52Phe) within the active site of the human proteasome. Specifically, we sought to understand how these mutations might disrupt protein function either by altering protein stability or by impeding interactions with a clinical administered drug. Leveraging molecular dynamics simulations and molecular docking calculations, we assessed the effect of these mutations on protein stability and ligand affinity. Notably, our results indicate that the Cys52Phe mutation critically impacts protein-ligand binding, providing valuable insights into potential proteasome inhibitor resistance.
Keywords: 20S proteasome inhibitors; drug resistance; molecular docking; molecular dynamics; mutations.
Copyright © 2024 Fernandes, Guedes, Victor, Salvador and Guedes.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
Similar articles
-
Exploring EZH2-Proteasome Dual-Targeting Drug Discovery through a Computational Strategy to Fight Multiple Myeloma.Molecules. 2021 Sep 14;26(18):5574. doi: 10.3390/molecules26185574. Molecules. 2021. PMID: 34577052 Free PMC article.
-
Probing the anticancer mechanism of prospective herbal drug Withaferin A on mammals: a case study on human and bovine proteasomes.BMC Genomics. 2010 Dec 2;11 Suppl 4(Suppl 4):S15. doi: 10.1186/1471-2164-11-S4-S15. BMC Genomics. 2010. PMID: 21143798 Free PMC article.
-
Support vector machine based prediction of P. falciparum proteasome inhibitors and development of focused library by molecular docking.Comb Chem High Throughput Screen. 2011 Dec;14(10):898-907. doi: 10.2174/138620711797537058. Comb Chem High Throughput Screen. 2011. PMID: 21843142
-
Updated Review and Perspective on 20S Proteasome Inhibitors in the Treatment of Lung Cancer.Curr Cancer Drug Targets. 2020;20(6):392-409. doi: 10.2174/1568009620666200226094000. Curr Cancer Drug Targets. 2020. PMID: 32101123 Review.
-
The cryo-EM structure of the Plasmodium falciparum 20S proteasome and its use in the fight against malaria.FEBS J. 2016 Dec;283(23):4238-4243. doi: 10.1111/febs.13780. Epub 2016 Jul 2. FEBS J. 2016. PMID: 27286897 Free PMC article. Review.
Cited by
-
Different Strategies to Overcome Resistance to Proteasome Inhibitors-A Summary 20 Years after Their Introduction.Int J Mol Sci. 2024 Aug 16;25(16):8949. doi: 10.3390/ijms25168949. Int J Mol Sci. 2024. PMID: 39201634 Free PMC article. Review.
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1 (2), 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., Hermans J. (1981). “Interaction models for water in relation to protein hydration,” in Intermolecular forces: proceedings of the fourteenth Jerusalem symposium on quantum Chemistry and biochemistry held in Jerusalem, Israel, april 13--16, 1981. Editor Pullman B. (Dordrecht: Springer Netherlands; ), 331–342. 10.1007/978-94-015-7658-1_21 - DOI
LinkOut - more resources
Full Text Sources