

Compartment-specific regulation of NaV1.7 in sensory neurons after acute exposure to TNF-α
- PMID: 38261513
- PMCID: PMC10947185
- DOI: 10.1016/j.celrep.2024.113685
Compartment-specific regulation of NaV1.7 in sensory neurons after acute exposure to TNF-α
Abstract
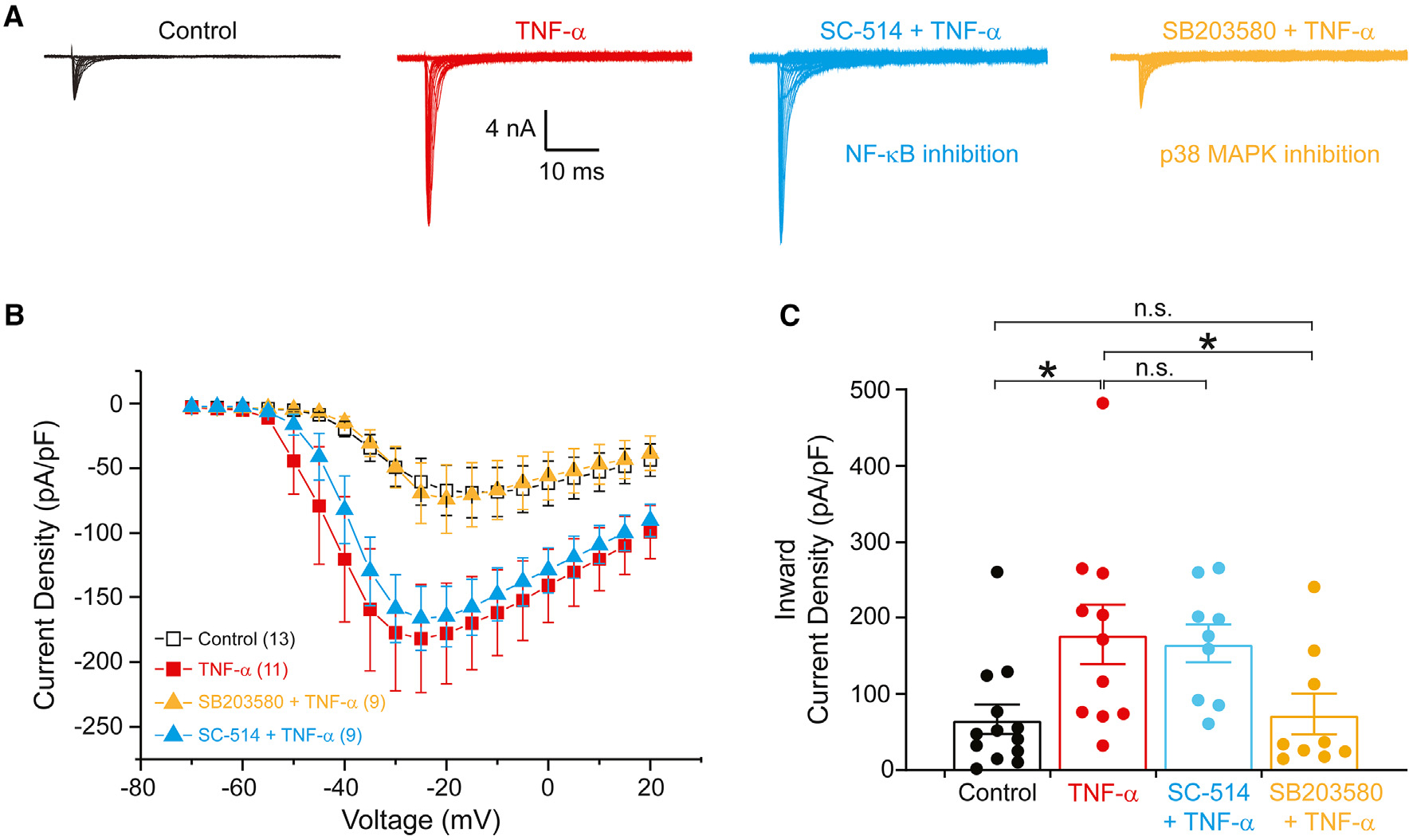
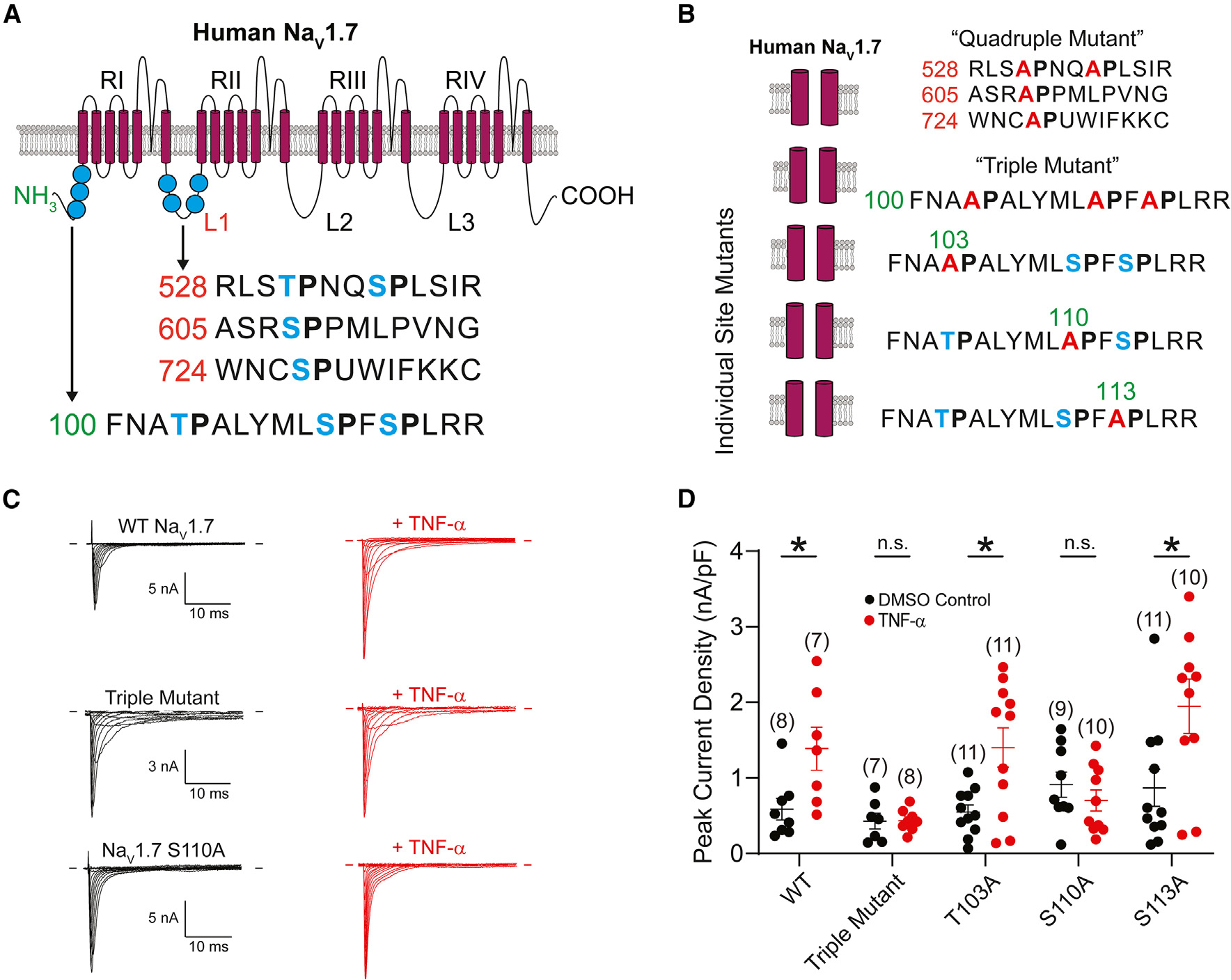
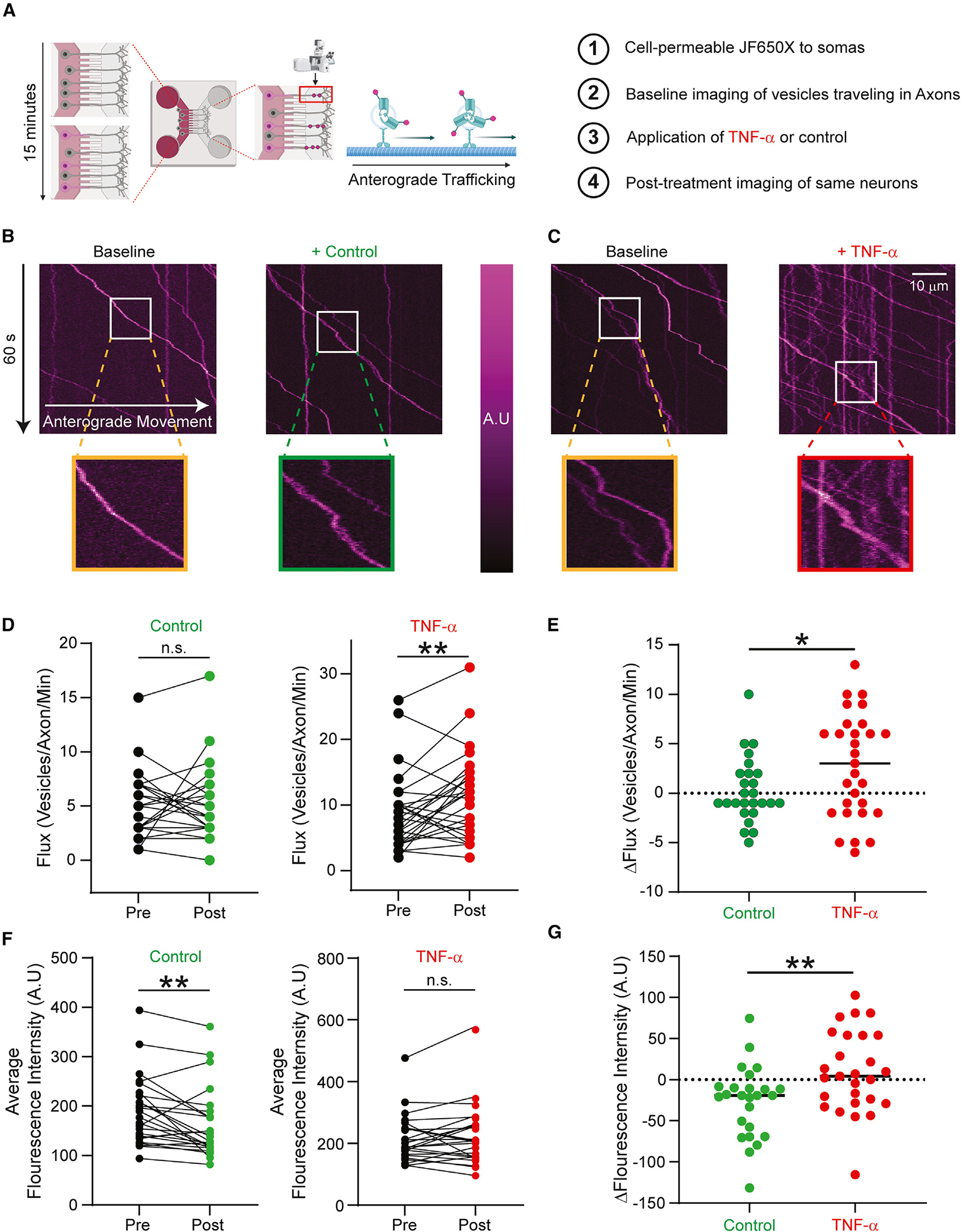
Tumor necrosis factor α (TNF-α) is a major pro-inflammatory cytokine, important in many diseases, that sensitizes nociceptors through its action on a variety of ion channels, including voltage-gated sodium (NaV) channels. We show here that TNF-α acutely upregulates sensory neuron excitability and current density of threshold channel NaV1.7. Using electrophysiological recordings and live imaging, we demonstrate that this effect on NaV1.7 is mediated by p38 MAPK and identify serine 110 in the channel's N terminus as the phospho-acceptor site, which triggers NaV1.7 channel insertion into the somatic membrane. We also show that the N terminus of NaV1.7 is sufficient to mediate this effect. Although acute TNF-α treatment increases NaV1.7-carrying vesicle accumulation at axonal endings, we did not observe increased channel insertion into the axonal membrane. These results identify molecular determinants of TNF-α-mediated regulation of NaV1.7 in sensory neurons and demonstrate compartment-specific effects of TNF-α on channel insertion in the neuronal plasma membrane.
Keywords: CP: Neuroscience; Na(V)1.7; TNF-α; distal axons; inflammatory pain; neuronal compartments; soma.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures




Similar articles
-
Chronic exposure to tumor necrosis factor in vivo induces hyperalgesia, upregulates sodium channel gene expression and alters the cellular electrophysiology of dorsal root ganglion neurons.Neurosci Lett. 2017 Jul 13;653:195-201. doi: 10.1016/j.neulet.2017.05.004. Epub 2017 May 27. Neurosci Lett. 2017. PMID: 28558976
-
Enhancement by TNF-α of TTX-resistant NaV current in muscle sensory neurons after femoral artery occlusion.Am J Physiol Regul Integr Comp Physiol. 2020 Apr 1;318(4):R772-R780. doi: 10.1152/ajpregu.00338.2019. Epub 2020 Feb 26. Am J Physiol Regul Integr Comp Physiol. 2020. PMID: 32101460 Free PMC article.
-
TNF-α acutely enhances acid-sensing ion channel currents in rat dorsal root ganglion neurons via a p38 MAPK pathway.J Neuroinflammation. 2021 Apr 14;18(1):92. doi: 10.1186/s12974-021-02151-w. J Neuroinflammation. 2021. PMID: 33853615 Free PMC article.
-
The Role of Voltage-Gated Sodium Channels in Pain Signaling.Physiol Rev. 2019 Apr 1;99(2):1079-1151. doi: 10.1152/physrev.00052.2017. Physiol Rev. 2019. PMID: 30672368 Review.
-
NaV1.9: a sodium channel linked to human pain.Nat Rev Neurosci. 2015 Sep;16(9):511-9. doi: 10.1038/nrn3977. Epub 2015 Aug 5. Nat Rev Neurosci. 2015. PMID: 26243570 Review.
Cited by
-
Nav1.8, an analgesic target for nonpsychotomimetic phytocannabinoids.Proc Natl Acad Sci U S A. 2025 Jan 28;122(4):e2416886122. doi: 10.1073/pnas.2416886122. Epub 2025 Jan 21. Proc Natl Acad Sci U S A. 2025. PMID: 39835903 Free PMC article.
-
The Role of TNF-α in Neuropathic Pain: An Immunotherapeutic Perspective.Life (Basel). 2025 May 14;15(5):785. doi: 10.3390/life15050785. Life (Basel). 2025. PMID: 40430212 Free PMC article. Review.
-
Worldwide Sodium Channel Conference, January 31st-February 2nd, 2024, Grindelwald, Switzerland.Bioelectricity. 2024 Dec 13;6(4):288-291. doi: 10.1089/bioe.2024.0025. eCollection 2024 Dec. Bioelectricity. 2024. PMID: 39712211
-
Neuronal p38 MAPK Signaling Contributes to Cisplatin-Induced Peripheral Neuropathy.Antioxidants (Basel). 2025 Apr 8;14(4):445. doi: 10.3390/antiox14040445. Antioxidants (Basel). 2025. PMID: 40298791 Free PMC article.
-
Unveiling the Mechanisms of Pain in Endometriosis: Comprehensive Analysis of Inflammatory Sensitization and Therapeutic Potential.Int J Mol Sci. 2025 Feb 19;26(4):1770. doi: 10.3390/ijms26041770. Int J Mol Sci. 2025. PMID: 40004233 Free PMC article. Review.
References
-
- Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F, and Strichartz GR (2006). The Role of Sodium Channels in Chronic Inflammatory and Neuropathic Pain. J. Pain 7. S1–29. - PubMed
-
- Kidd BL, and Urban LA (2001). Mechanisms of inflammatory pain. Br. J. Anaesth. 87, 3–11. - PubMed
-
- Talbot S, Foster SL, and Woolf CJ (2016). Neuroimmunity: Physiology and Pathology. Annu. Rev. Immunol. 34, 421–447. - PubMed
-
- Bennett DL, Clark AJ, Huang J, Waxman SG, and Dib-Hajj SD (2019). The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 99, 1079–1151. - PubMed
-
- Dib-Hajj SD, Yang Y, Black JA, and Waxman SG (2013). The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
