

Loss of Airway Phylogenetic Diversity Is Associated with Clinical and Pathobiological Markers of Disease Development in Chronic Obstructive Pulmonary Disease
- PMID: 38261629
- PMCID: PMC11273318
- DOI: 10.1164/rccm.202303-0489OC
Loss of Airway Phylogenetic Diversity Is Associated with Clinical and Pathobiological Markers of Disease Development in Chronic Obstructive Pulmonary Disease
Abstract
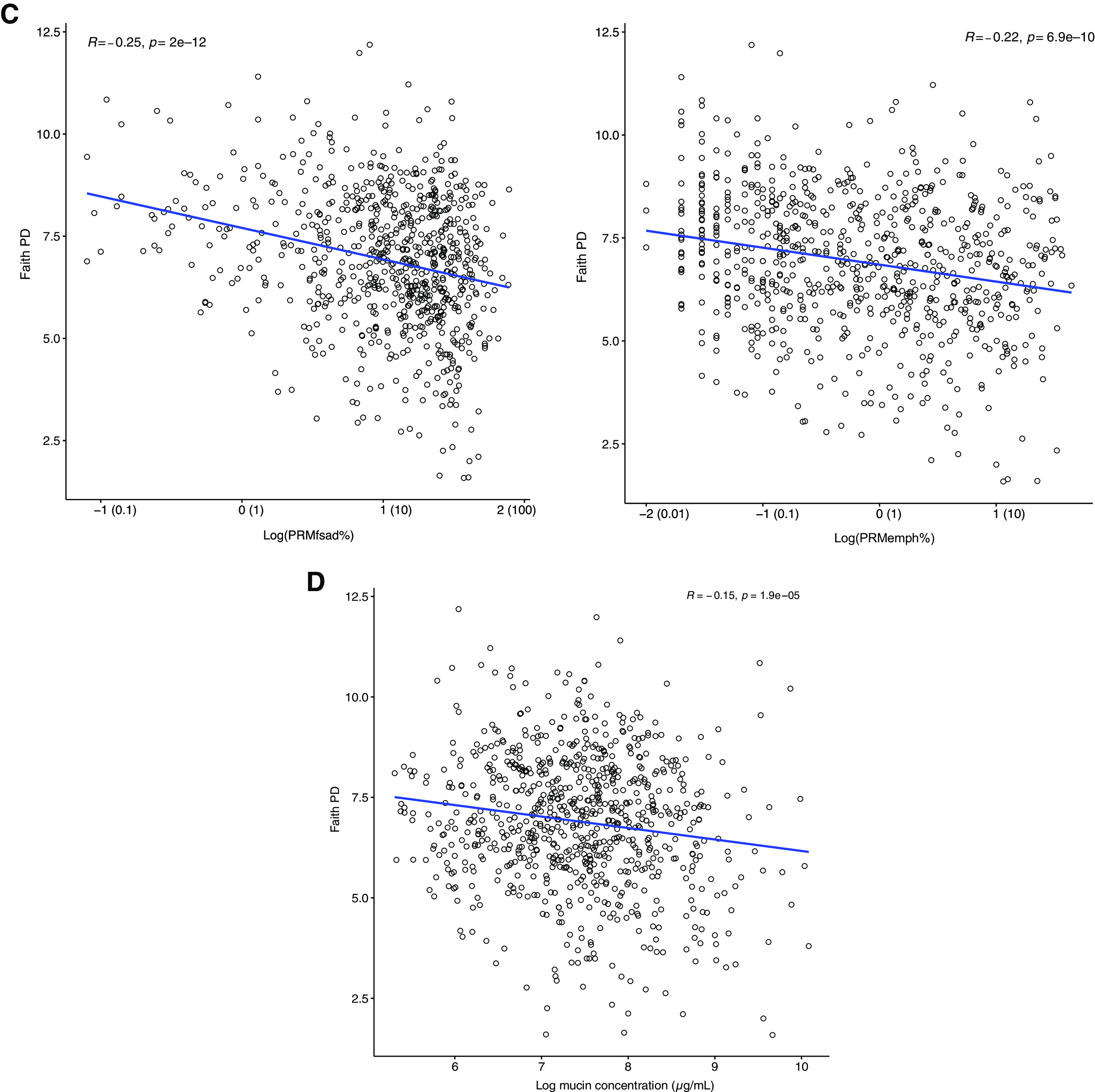
Rationale: The airway microbiome has the potential to shape chronic obstructive pulmonary disease (COPD) pathogenesis, but its relationship to outcomes in milder disease is unestablished. Objectives: To identify sputum microbiome characteristics associated with markers of COPD in participants of the Subpopulations and Intermediate Outcome Measures of COPD Study (SPIROMICS). Methods: Sputum DNA from 877 participants was analyzed using 16S ribosomal RNA gene sequencing. Relationships between baseline airway microbiota composition and clinical, radiographic, and mucoinflammatory markers, including longitudinal lung function trajectory, were examined. Measurements and Main Results: Participant data represented predominantly milder disease (Global Initiative for Chronic Obstructive Lung Disease stage 0-2 obstruction in 732 of 877 participants). Phylogenetic diversity (i.e., range of different species within a sample) correlated positively with baseline lung function, decreased with higher Global Initiative for Chronic Obstructive Lung Disease stage, and correlated negatively with symptom burden, radiographic markers of airway disease, and total mucin concentrations (P < 0.001). In covariate-adjusted regression models, organisms robustly associated with better lung function included Alloprevotella, Oribacterium, and Veillonella species. Conversely, lower lung function, greater symptoms, and radiographic measures of small airway disease were associated with enrichment in members of Streptococcus, Actinobacillus, Actinomyces, and other genera. Baseline sputum microbiota features were also associated with lung function trajectory during SPIROMICS follow-up (stable/improved, decline, or rapid decline groups). The stable/improved group (slope of FEV1 regression ⩾66th percentile) had greater bacterial diversity at baseline associated with enrichment in Prevotella, Leptotrichia, and Neisseria species. In contrast, the rapid decline group (FEV1 slope ⩽33rd percentile) had significantly lower baseline diversity associated with enrichment in Streptococcus species. Conclusions: In SPIROMICS, baseline airway microbiota features demonstrate divergent associations with better or worse COPD-related outcomes.
Keywords: lung function; microbiome; microbiota; mucin; sputum.
Figures






Comment in
-
Unsung Heroes? Decoding the Protective Effects of Airway Microbiota in Chronic Obstructive Pulmonary Disease.Am J Respir Crit Care Med. 2024 Jul 15;210(2):136-138. doi: 10.1164/rccm.202401-0189ED. Am J Respir Crit Care Med. 2024. PMID: 38358821 Free PMC article. No abstract available.
References
-
- Mammen MJ, Sethi S. COPD and the microbiome. Respirology . 2016;21:590–599. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HHSN268200900019C/HL/NHLBI NIH HHS/United States
- P30 ES005605/ES/NIEHS NIH HHS/United States
- R01 HL121774/HL/NHLBI NIH HHS/United States
- K24 HL137013/HL/NHLBI NIH HHS/United States
- HHSN268200900015C/HL/NHLBI NIH HHS/United States
- HHSN268200900016C/HL/NHLBI NIH HHS/United States
- U01 HL137880/HL/NHLBI NIH HHS/United States
- HHSN268200900013C/HL/NHLBI NIH HHS/United States
- R01 AI129958/AI/NIAID NIH HHS/United States
- HHSN268200900014C/HL/NHLBI NIH HHS/United States
- U24 HL141762/HL/NHLBI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- HHSN268200900018C/HL/NHLBI NIH HHS/United States
- P30 DK054759/DK/NIDDK NIH HHS/United States
- HHSN268200900017C/HL/NHLBI NIH HHS/United States
- HHSN268200900020C/HL/NHLBI NIH HHS/United States
- R01 HL144718/HL/NHLBI NIH HHS/United States
- K08 HL157651/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
