

A novel MTORC2-AKT-ROS axis triggers mitofission and mitophagy-associated execution of colorectal cancer cells upon drug-induced activation of mutant KRAS
- PMID: 38261660
- PMCID: PMC11210925
- DOI: 10.1080/15548627.2024.2307224
A novel MTORC2-AKT-ROS axis triggers mitofission and mitophagy-associated execution of colorectal cancer cells upon drug-induced activation of mutant KRAS
Abstract
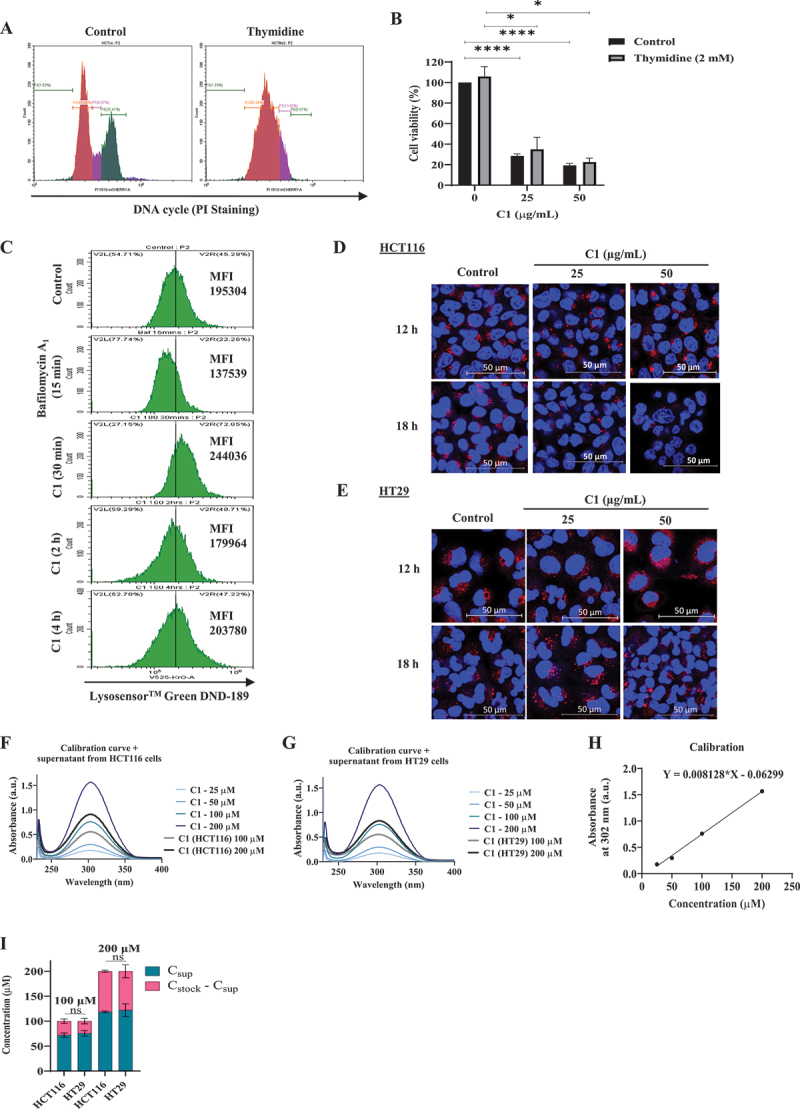
RAS is one of the most commonly mutated oncogenes associated with multiple cancer hallmarks. Notably, RAS activation induces intracellular reactive oxygen species (ROS) generation, which we previously demonstrated as a trigger for autophagy-associated execution of mutant KRAS-expressing cancer cells. Here we report that drug (merodantoin; C1)-induced activation of mutant KRAS promotes phospho-AKT S473-dependent ROS-mediated S616 phosphorylation and mitochondrial localization of DNM1L/DRP1 (dynamin 1 like) and cleavage of the fusion-associated protein OPA1 (OPA1 mitochondrial dynamin like GTPase). Interestingly, accumulation of the outer mitochondrial membrane protein VDAC1 (voltage dependent anion channel 1) is observed in mutant KRAS-expressing cells upon exposure to C1. Conversely, silencing VDAC1 abolishes C1-induced mitophagy, and gene knockdown of either KRAS, AKT or DNM1L rescues ROS-dependent VDAC1 accumulation and stability, thus suggesting an axis of mutant active KRAS-phospho-AKT S473-ROS-DNM1L-VDAC1 in mitochondrial morphology change and cancer cell execution. Importantly, we identified MTOR (mechanistic target of rapamycin kinsase) complex 2 (MTORC2) as the upstream mediator of AKT phosphorylation at S473 in our model. Pharmacological or genetic inhibition of MTORC2 abrogated C1-induced phosphorylation of AKT S473, ROS generation and mitophagy induction, as well as rescued tumor colony forming ability and migratory capacity. Finally, increase in thermal stability of KRAS, AKT and DNM1L were observed upon exposure to C1 only in mutant KRAS-expressing cells. Taken together, our work has unraveled a novel mechanism of selective targeting of mutant KRAS-expressing cancers via MTORC2-mediated AKT activation and ROS-dependent mitofission, which could have potential therapeutic implications given the relative lack of direct RAS-targeting strategies in cancer.Abbreviations: ACTB/ß-actin: actin beta; AKT: AKT serine/threonine kinase; C1/merodantoin: 1,3-dibutyl-2-thiooxo-imidazoldine-4,5-dione; CAT: catalase; CETSA: cellular thermal shift assay; CHX: cycloheximide; DKO: double knockout; DNM1L/DRP1: dynamin 1 like; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; H2O2: hydrogen peroxide; HSPA1A/HSP70-1: heat shock protein family A (Hsp70) member 1A; HSP90AA1/HSP90: heat shock protein 90 alpha family class A member 1; KRAS: KRAS proto-oncogene, GTPase; MAP1LC3B/LC3B, microtubule associated protein 1 light chain 3 beta; LC3B-I: unlipidated form of LC3B; LC3B-II: phosphatidylethanolamine-conjugated form of LC3B; MAPKAP1/SIN1: MAPK associated protein 1; MAPK1/ERK2: mitogen-activated protein kinase 1; MAPK3/ERK1: mitogen-activated protein kinase 3; MFI: mean fluorescence intensity; MiNA: Mitochondrial Network Analysis; MTOR: mechanistic target of rapamycin kinase; MTORC1: mechanistic target of rapamycin kinase complex 1; MTORC2: mechanistic target of rapamycin kinase complex 2; O2.-: superoxide; OMA1: OMA1 zinc metallopeptidase; OPA1: OPA1 mitochondrial dynamin like GTPase; RICTOR: RPTOR independent companion of MTOR complex 2; ROS: reactive oxygen species; RPTOR/raptor: regulatory associated protein of MTOR complex 1; SOD1: superoxide dismutase 1; SOD2: superoxide dismutase 2; SQSTM1/p62: sequestosome 1; VDAC1: voltage dependent anion channel 1; VDAC2: voltage dependent anion channel 2.
Keywords: AKT; DNM1L; KRAS; MTORC2; ROS; mitofission.
Conflict of interest statement
Some of the data presented in the manuscript are part of the doctoral thesis of Jonathan Foo, NUS Graduate School and Department of Physiology, YLL School of Medicine, National University of Singapore, Singapore.
Figures








Similar articles
-
Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation.Autophagy. 2019 Sep;15(9):1606-1619. doi: 10.1080/15548627.2019.1591672. Epub 2019 Apr 7. Autophagy. 2019. PMID: 30859901 Free PMC article.
-
Autophagy drives fibroblast senescence through MTORC2 regulation.Autophagy. 2020 Nov;16(11):2004-2016. doi: 10.1080/15548627.2020.1713640. Epub 2020 Jan 13. Autophagy. 2020. PMID: 31931659 Free PMC article.
-
Aberrant mitochondrial morphology and function associated with impaired mitophagy and DNM1L-MAPK/ERK signaling are found in aged mutant Parkinsonian LRRK2R1441G mice.Autophagy. 2021 Oct;17(10):3196-3220. doi: 10.1080/15548627.2020.1850008. Epub 2020 Dec 10. Autophagy. 2021. PMID: 33300446 Free PMC article.
-
Autophagy in the physiological endometrium and cancer.Autophagy. 2021 May;17(5):1077-1095. doi: 10.1080/15548627.2020.1752548. Epub 2020 May 13. Autophagy. 2021. PMID: 32401642 Free PMC article. Review.
-
Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles.Autophagy. 2021 Feb;17(2):385-401. doi: 10.1080/15548627.2020.1725377. Epub 2020 Feb 12. Autophagy. 2021. PMID: 32048886 Free PMC article. Review.
Cited by
-
Acetylated Dendrobium huoshanense polysaccharide: a novel inducer of apoptosis in colon cancer cells via Fas-FasL pathway activation and metabolic reprogramming.Front Oncol. 2025 Mar 4;15:1529868. doi: 10.3389/fonc.2025.1529868. eCollection 2025. Front Oncol. 2025. PMID: 40104499 Free PMC article.
-
A Mitochondria-Related Signature in Diffuse Large B-Cell Lymphoma: Prognosis, Immune and Therapeutic Features.Cancer Med. 2025 Jan;14(2):e70602. doi: 10.1002/cam4.70602. Cancer Med. 2025. PMID: 39811936 Free PMC article.
-
Interplay between epigenetics, senescence and cellular redox metabolism in cancer and its therapeutic implications.Redox Biol. 2024 Dec;78:103441. doi: 10.1016/j.redox.2024.103441. Epub 2024 Nov 23. Redox Biol. 2024. PMID: 39612910 Free PMC article. Review.
-
Mitochondrial metabolism and cancer therapeutic innovation.Signal Transduct Target Ther. 2025 Aug 4;10(1):245. doi: 10.1038/s41392-025-02311-x. Signal Transduct Target Ther. 2025. PMID: 40754534 Free PMC article. Review.
-
Mitochondrial Reactive Oxygen Species (mROS) Generation and Cancer: Emerging Nanoparticle Therapeutic Approaches.Int J Nanomedicine. 2025 May 13;20:6085-6119. doi: 10.2147/IJN.S510972. eCollection 2025. Int J Nanomedicine. 2025. PMID: 40385494 Free PMC article. Review.
References
-
- Jänne PA, Rybkin II, Spira AI, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in Advanced/Metastatic Non Small-Cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur J Cancer. 2020;138:S1–S2. doi: 10.1016/S0959-8049(20)31076-5 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous