

Yeast eIF2A has a minimal role in translation initiation and uORF-mediated translational control in vivo
- PMID: 38266075
- PMCID: PMC10945734
- DOI: 10.7554/eLife.92916
Yeast eIF2A has a minimal role in translation initiation and uORF-mediated translational control in vivo
Abstract
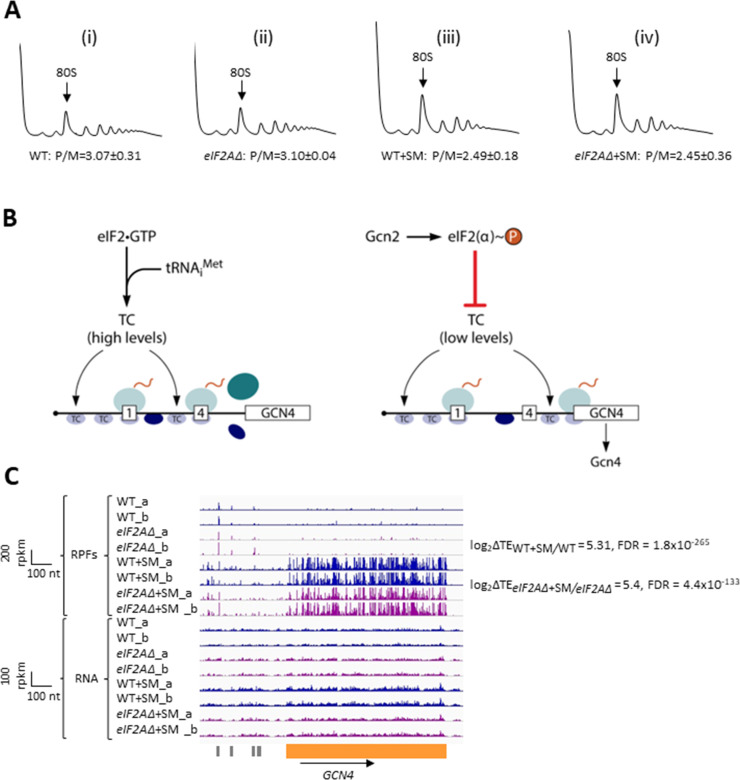
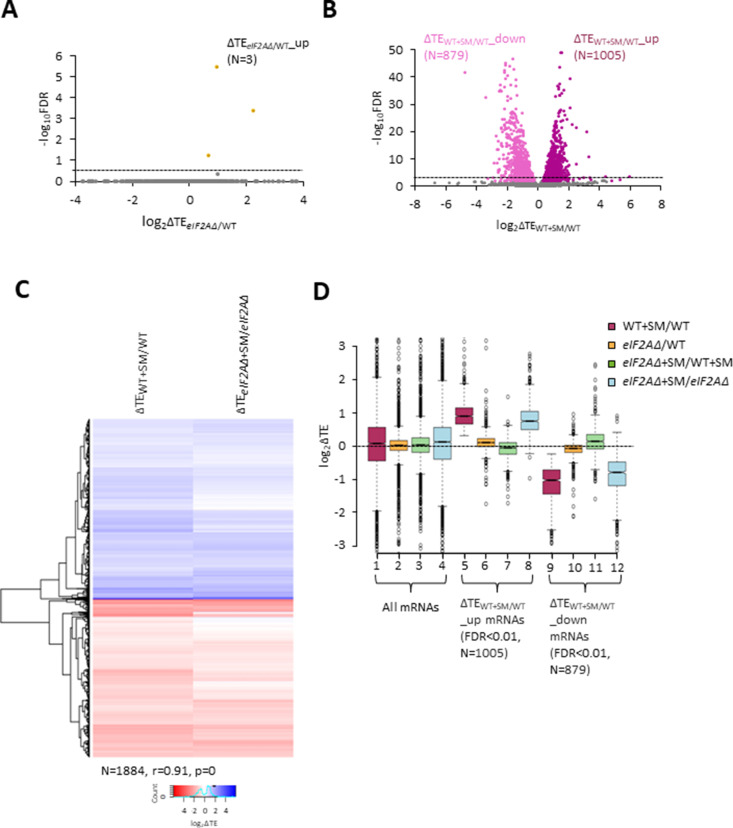
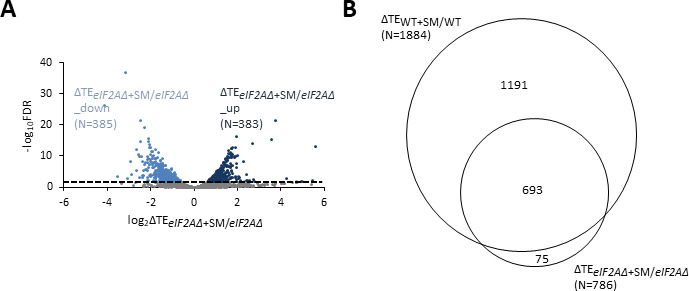
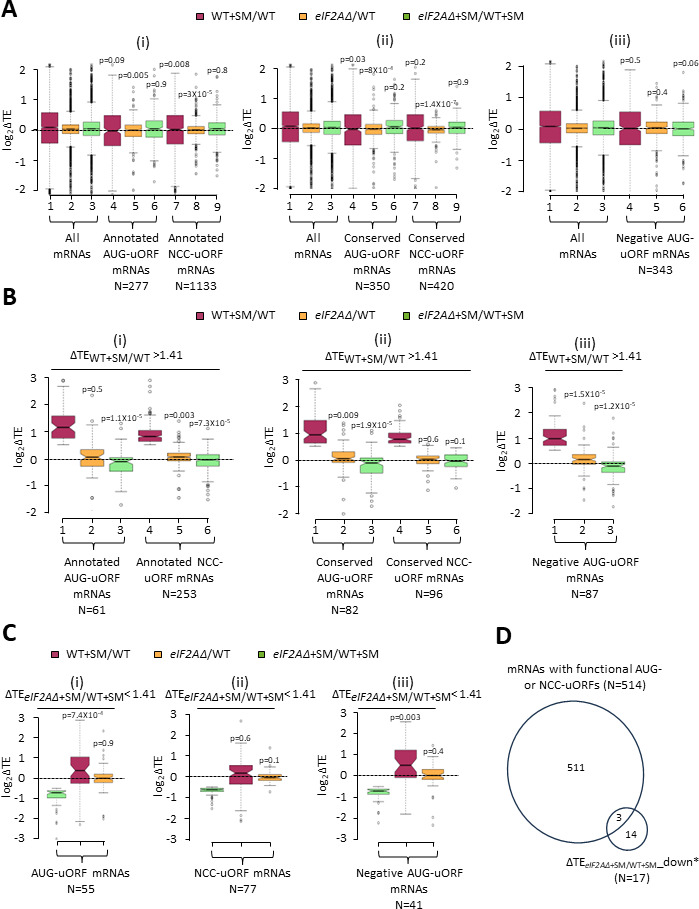
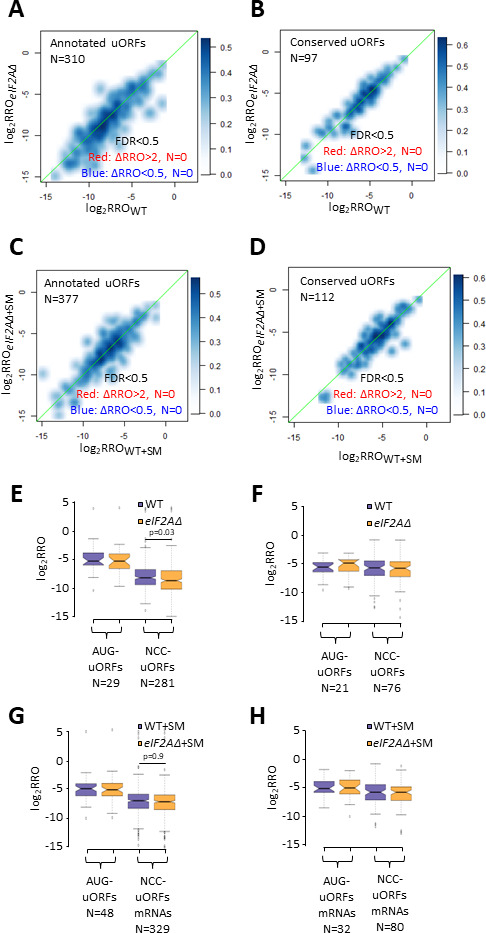
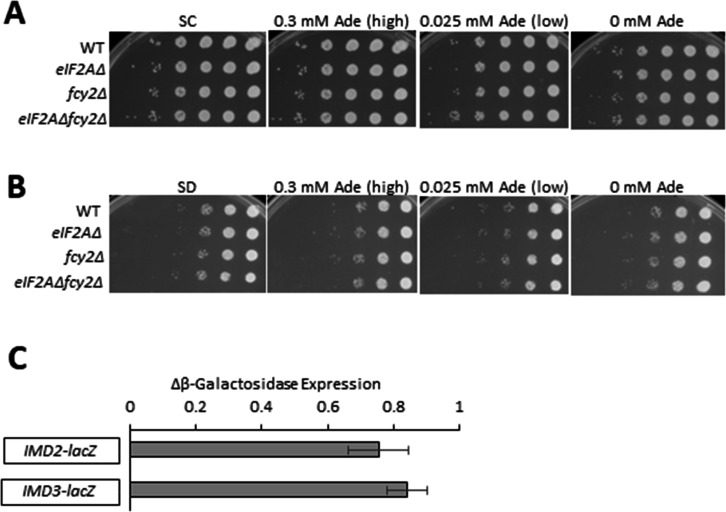
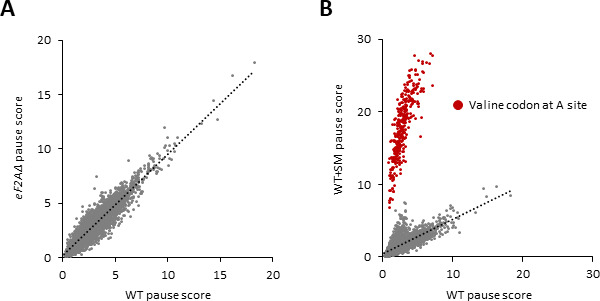
Initiating translation of most eukaryotic mRNAs depends on recruitment of methionyl initiator tRNA (Met-tRNAi) in a ternary complex (TC) with GTP-bound eukaryotic initiation factor 2 (eIF2) to the small (40S) ribosomal subunit, forming a 43S preinitiation complex (PIC) that attaches to the mRNA and scans the 5'-untranslated region (5' UTR) for an AUG start codon. Previous studies have implicated mammalian eIF2A in GTP-independent binding of Met-tRNAi to the 40S subunit and its recruitment to specialized mRNAs that do not require scanning, and in initiation at non-AUG start codons, when eIF2 function is attenuated by phosphorylation of its α-subunit during stress. The role of eIF2A in translation in vivo is poorly understood however, and it was unknown whether the conserved ortholog in budding yeast can functionally substitute for eIF2. We performed ribosome profiling of a yeast deletion mutant lacking eIF2A and isogenic wild-type (WT) cells in the presence or absence of eIF2α phosphorylation induced by starvation for amino acids isoleucine and valine. Whereas starvation of WT confers changes in translational efficiencies (TEs) of hundreds of mRNAs, the eIF2AΔ mutation conferred no significant TE reductions for any mRNAs in non-starved cells, and it reduced the TEs of only a small number of transcripts in starved cells containing phosphorylated eIF2α. We found no evidence that eliminating eIF2A altered the translation of mRNAs containing putative internal ribosome entry site (IRES) elements, or harboring uORFs initiated by AUG or near-cognate start codons, in non-starved or starved cells. Thus, very few mRNAs (possibly only one) appear to employ eIF2A for Met-tRNAi recruitment in yeast cells, even when eIF2 function is attenuated by stress.
Keywords: IRES; S. cerevisiae; eIF2; eIF2A; genetics; genomics; translation; uORF; yeast.
Conflict of interest statement
SG, FG, HZ No competing interests declared, AH Reviewing editor, eLife
Figures
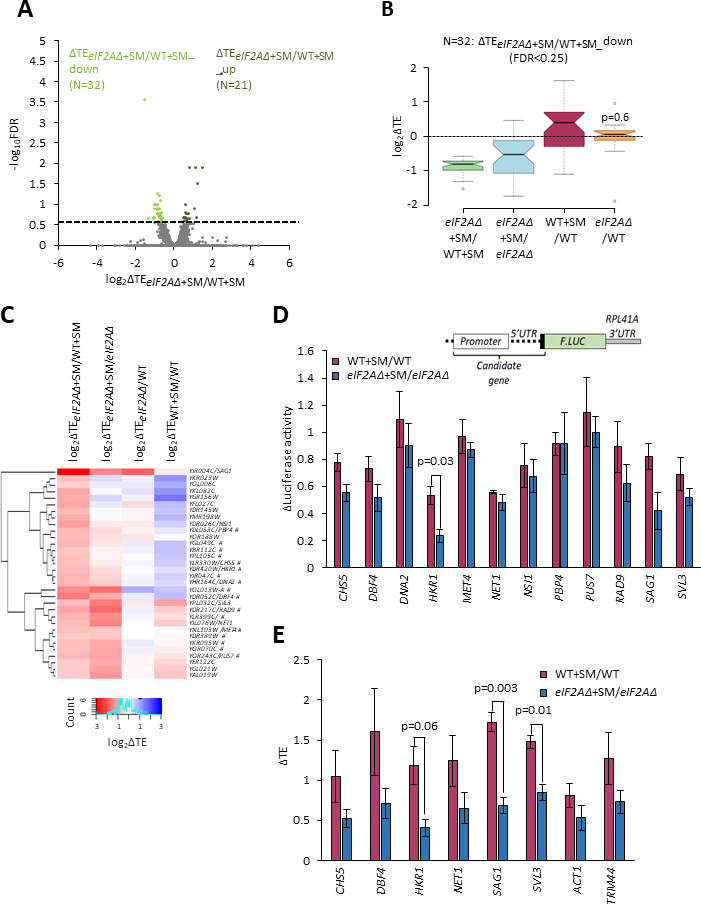
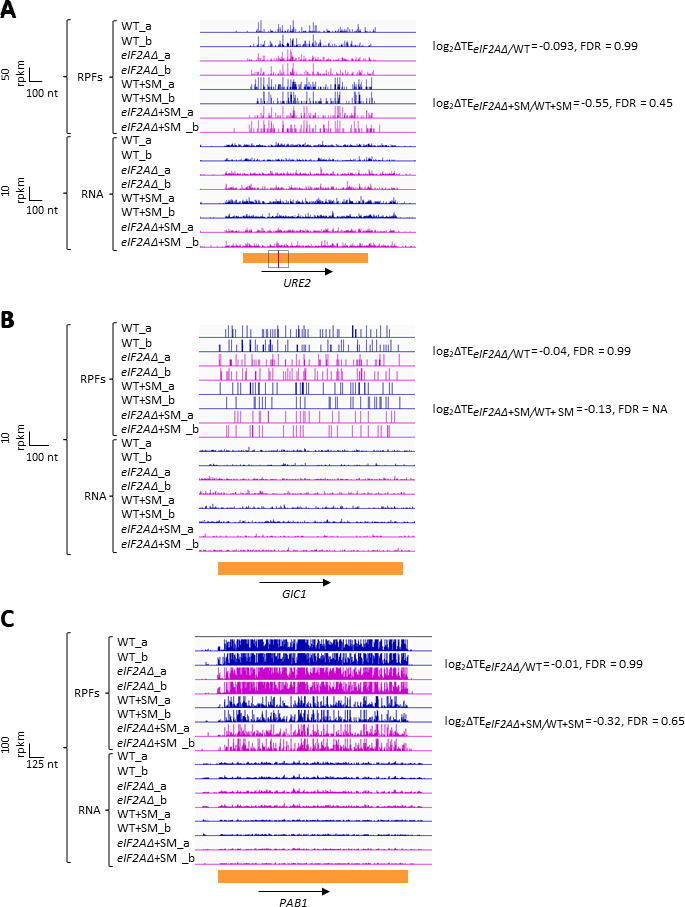
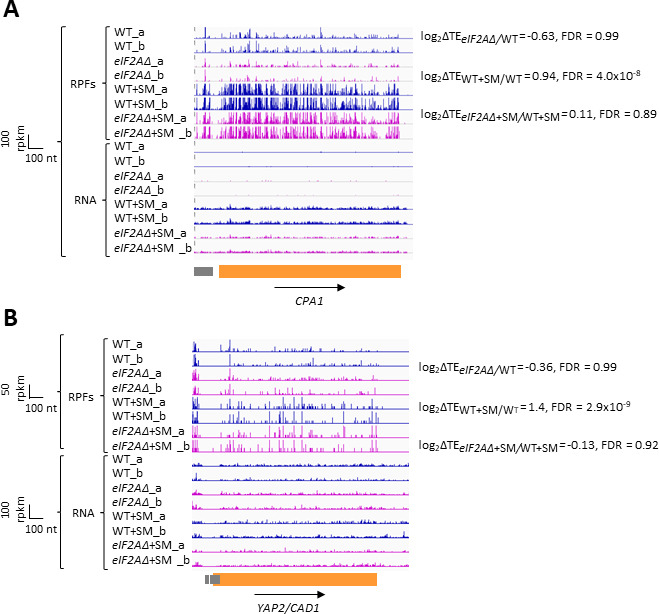
Update of
-
Yeast eIF2A has a minimal role in translation initiation and uORF-mediated translational control in vivo.bioRxiv [Preprint]. 2023 Dec 9:2023.10.06.561292. doi: 10.1101/2023.10.06.561292. bioRxiv. 2023. Update in: Elife. 2024 Jan 24;12:RP92916. doi: 10.7554/eLife.92916. PMID: 37986989 Free PMC article. Updated. Preprint.
Similar articles
-
Yeast eIF2A has a minimal role in translation initiation and uORF-mediated translational control in vivo.bioRxiv [Preprint]. 2023 Dec 9:2023.10.06.561292. doi: 10.1101/2023.10.06.561292. bioRxiv. 2023. Update in: Elife. 2024 Jan 24;12:RP92916. doi: 10.7554/eLife.92916. PMID: 37986989 Free PMC article. Updated. Preprint.
-
eIF1 Loop 2 interactions with Met-tRNAi control the accuracy of start codon selection by the scanning preinitiation complex.Proc Natl Acad Sci U S A. 2018 May 1;115(18):E4159-E4168. doi: 10.1073/pnas.1800938115. Epub 2018 Apr 16. Proc Natl Acad Sci U S A. 2018. PMID: 29666249 Free PMC article.
-
The eIF2A knockout mouse.Cell Cycle. 2016 Nov 16;15(22):3115-3120. doi: 10.1080/15384101.2016.1237324. Epub 2016 Sep 29. Cell Cycle. 2016. PMID: 27686860 Free PMC article.
-
Exploring the interaction dynamics of eukaryotic translation initiation factor 2.Biochem Soc Trans. 2025 Jun 30;53(3):593-602. doi: 10.1042/BST20253022. Biochem Soc Trans. 2025. PMID: 40411218 Free PMC article. Review.
-
A Retrospective on eIF2A-and Not the Alpha Subunit of eIF2.Int J Mol Sci. 2020 Mar 17;21(6):2054. doi: 10.3390/ijms21062054. Int J Mol Sci. 2020. PMID: 32192132 Free PMC article. Review.
Cited by
-
To initiate or not to initiate: A critical assessment of eIF2A, eIF2D, and MCT-1·DENR to deliver initiator tRNA to ribosomes.Wiley Interdiscip Rev RNA. 2024 Mar-Apr;15(2):e1833. doi: 10.1002/wrna.1833. Wiley Interdiscip Rev RNA. 2024. PMID: 38433101 Free PMC article. Review.
-
Eukaryotic translation initiation factor 2A protects pancreatic beta cells during endoplasmic reticulum stress while rescuing global translation inhibition.Diabetologia. 2025 Aug;68(8):1735-1753. doi: 10.1007/s00125-025-06431-5. Epub 2025 Apr 30. Diabetologia. 2025. PMID: 40304759
-
Human eIF2A has a minimal role in translation initiation and in uORF-mediated translational control in HeLa cells.Elife. 2025 Jul 2;14:RP105311. doi: 10.7554/eLife.105311. Elife. 2025. PMID: 40600802 Free PMC article.
-
eIF2A regulates cell migration in a translation-independent manner.Sci Adv. 2025 Aug;11(31):eadu5668. doi: 10.1126/sciadv.adu5668. Epub 2025 Aug 1. Sci Adv. 2025. PMID: 40749049 Free PMC article.
References
-
- Anderson R, Agarwal A, Ghosh A, Guan B-J, Casteel J, Dvorina N, Baldwin WM, Mazumder B, Nazarko TY, Merrick WC, Buchner DA, Hatzoglou M, Kondratov RV, Komar AA. eIF2A-knockout mice reveal decreased life span and metabolic syndrome. FASEB Journal. 2021;35:e21990. doi: 10.1096/fj.202101105R. - DOI - PMC - PubMed
-
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin ZY, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T, Bahr S, Chen Y, Deshpande R, Kurat CF, Li SC, Li Z, Usaj MM, Okada H, Pascoe N, San Luis BJ, Sharifpoor S, Shuteriqi E, Simpkins SW, Snider J, Suresh HG, Tan Y, Zhu H, Malod-Dognin N, Janjic V, Przulj N, Troyanskaya OG, Stagljar I, Xia T, Ohya Y, Gingras AC, Raught B, Boutros M, Steinmetz LM, Moore CL, Rosebrock AP, Caudy AA, Myers CL, Andrews B, Boone C. A global genetic interaction network maps A wiring diagram of cellular function. Science. 2016;353:aaf1420. doi: 10.1126/science.aaf1420. - DOI - PMC - PubMed
-
- Delbecq P, Werner M, Feller A, Filipkowski RK, Messenguy F, Piérard A. A segment of mRNA encoding the leader peptide of the CPA1 gene confers repression by arginine on A heterologous yeast gene transcript. Molecular and Cellular Biology. 1994;14:2378–2390. doi: 10.1128/mcb.14.4.2378-2390.1994. - DOI - PMC - PubMed
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
