

Increased dosage of DYRK1A leads to congenital heart defects in a mouse model of Down syndrome
- PMID: 38266108
- PMCID: PMC7615651
- DOI: 10.1126/scitranslmed.add6883
Increased dosage of DYRK1A leads to congenital heart defects in a mouse model of Down syndrome
Abstract
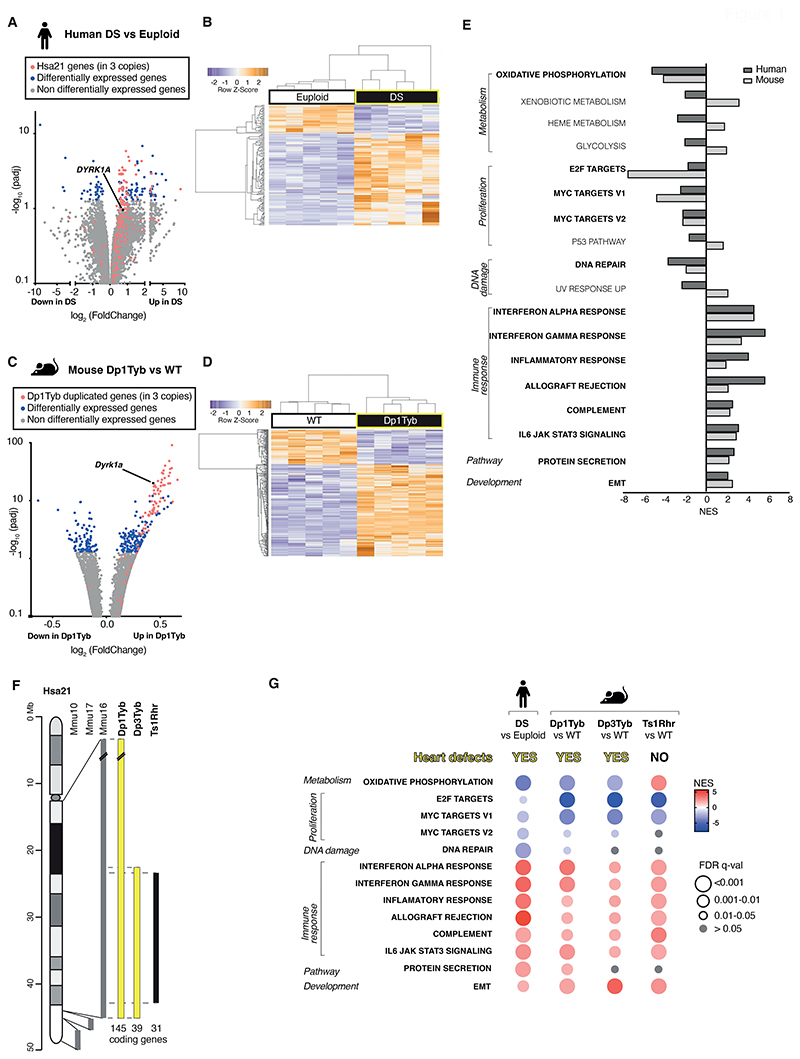
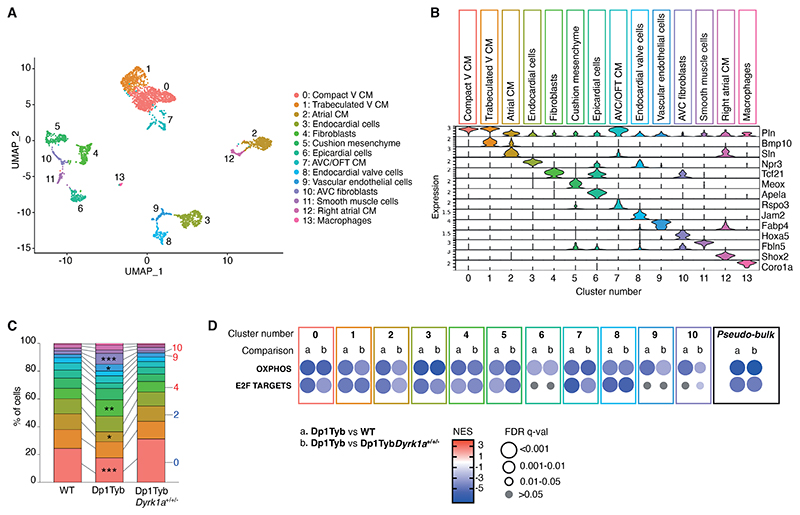
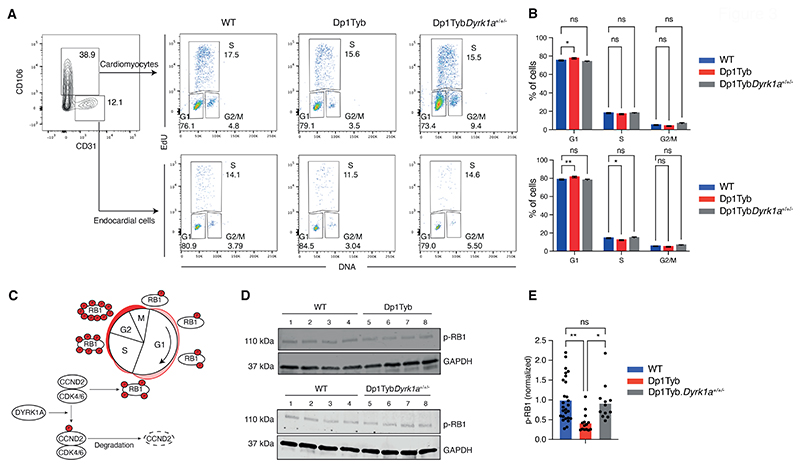
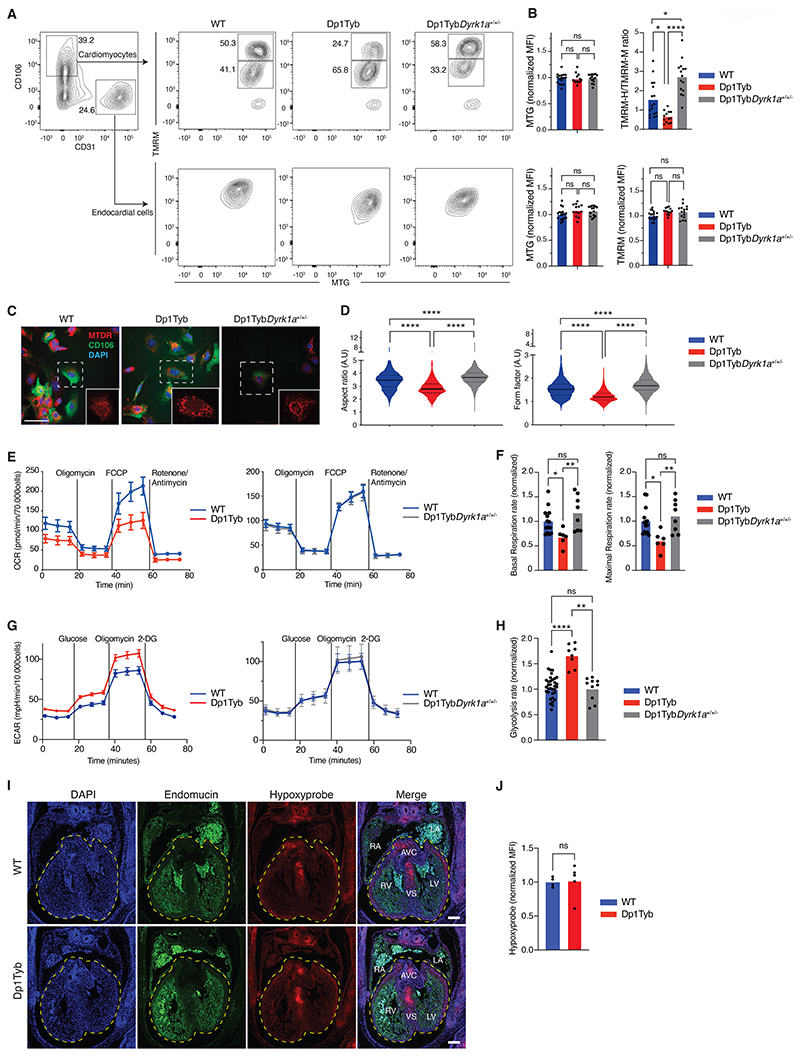
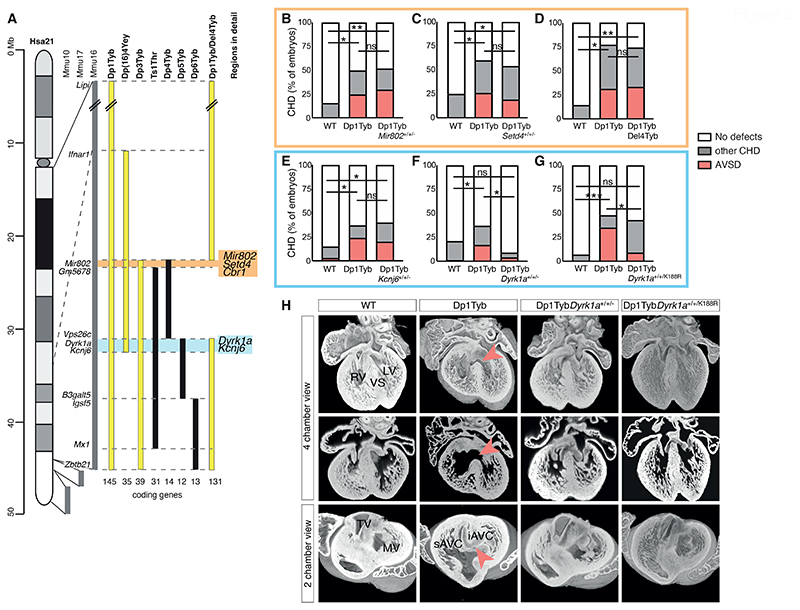
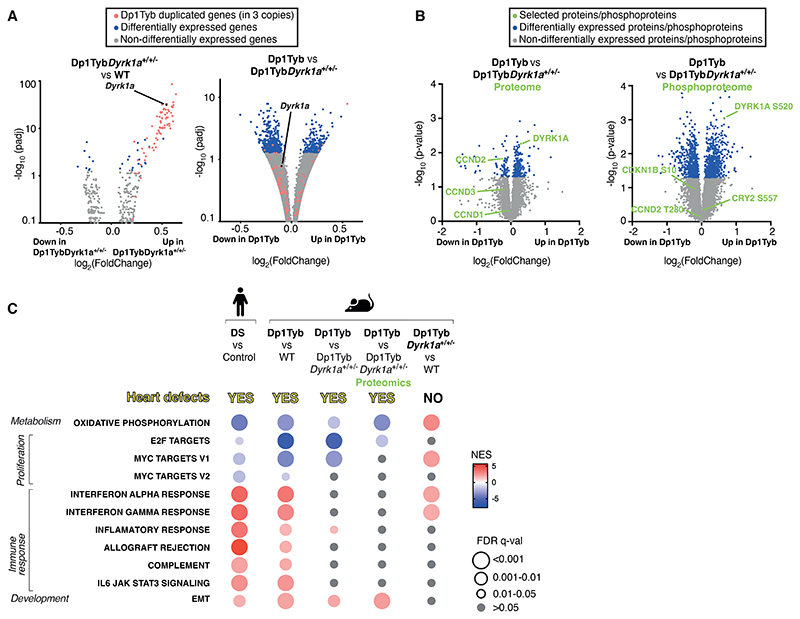
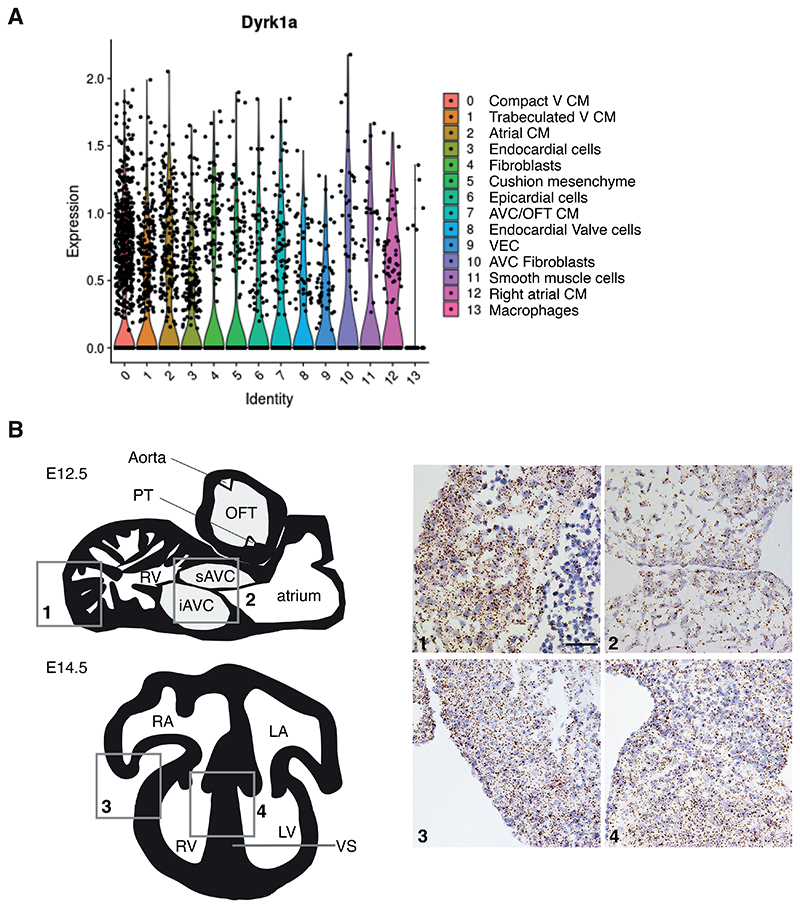
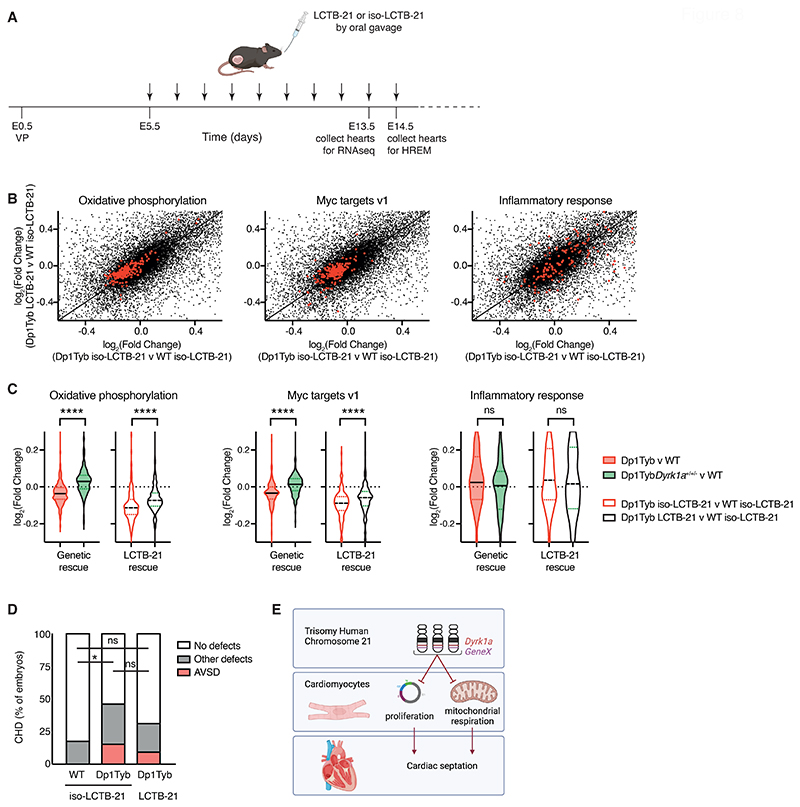
Down syndrome (DS) is caused by trisomy of human chromosome 21 (Hsa21). DS is a gene dosage disorder that results in multiple phenotypes including congenital heart defects. This clinically important cardiac pathology is the result of a third copy of one or more of the approximately 230 genes on Hsa21, but the identity of the causative dosage-sensitive genes and hence mechanisms underlying this cardiac pathology remain unclear. Here, we show that hearts from human fetuses with DS and embryonic hearts from the Dp1Tyb mouse model of DS show reduced expression of mitochondrial respiration genes and cell proliferation genes. Using systematic genetic mapping, we determined that three copies of the dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1a) gene, encoding a serine/threonine protein kinase, are associated with congenital heart disease pathology. In embryos from Dp1Tyb mice, reducing Dyrk1a gene copy number from three to two reversed defects in cellular proliferation and mitochondrial respiration in cardiomyocytes and rescued heart septation defects. Increased dosage of DYRK1A protein resulted in impairment of mitochondrial function and congenital heart disease pathology in mice with DS, suggesting that DYRK1A may be a useful therapeutic target for treating this common human condition.
Conflict of interest statement
L.M. is a founder of Perha Pharmaceuticals. L.M. and E.M. are co-inventors in the Leucettinibs patents (New imidazolone derivatives as inhibitors of protein kinases, in particular DYRK1A, CLK1 and/or CLK4; EP4143185, PCT/EP2021/061349, WO2021219828A1, EP4173675A1, EP4173673, EP4173674). All other authors have no competing interests.
Figures
Comment in
-
DYRK1A gene linked to heart defects in Down syndrome.Nat Rev Cardiol. 2024 Apr;21(4):217. doi: 10.1038/s41569-024-00995-2. Nat Rev Cardiol. 2024. PMID: 38326439 No abstract available.
References
-
- Freeman SB, Bean LH, Allen EG, Tinker SW, Locke AE, Druschel C, Hobbs CA, Romitti PA, Royle MH, Torfs CP, Dooley KJ, et al. Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down Syndrome Project. Genet Med. 2008;10:173–80. - PubMed
-
- Santoro SL, Steffensen EH. Congenital heart disease in Down syndrome – A review of temporal changes. Journal of Congenital Cardiology. 2021;5:1.
-
- Vis JC, Duffels MG, Winter MM, Weijerman ME, Cobben JM, Huisman SA, Mulder BJ. Down syndrome: a cardiovascular perspective. J Intellect Disabil Res. 2009;53:419–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
