

Oligodendrocyte-axon metabolic coupling is mediated by extracellular K+ and maintains axonal health
- PMID: 38267524
- PMCID: PMC10917689
- DOI: 10.1038/s41593-023-01558-3
Oligodendrocyte-axon metabolic coupling is mediated by extracellular K+ and maintains axonal health
Abstract
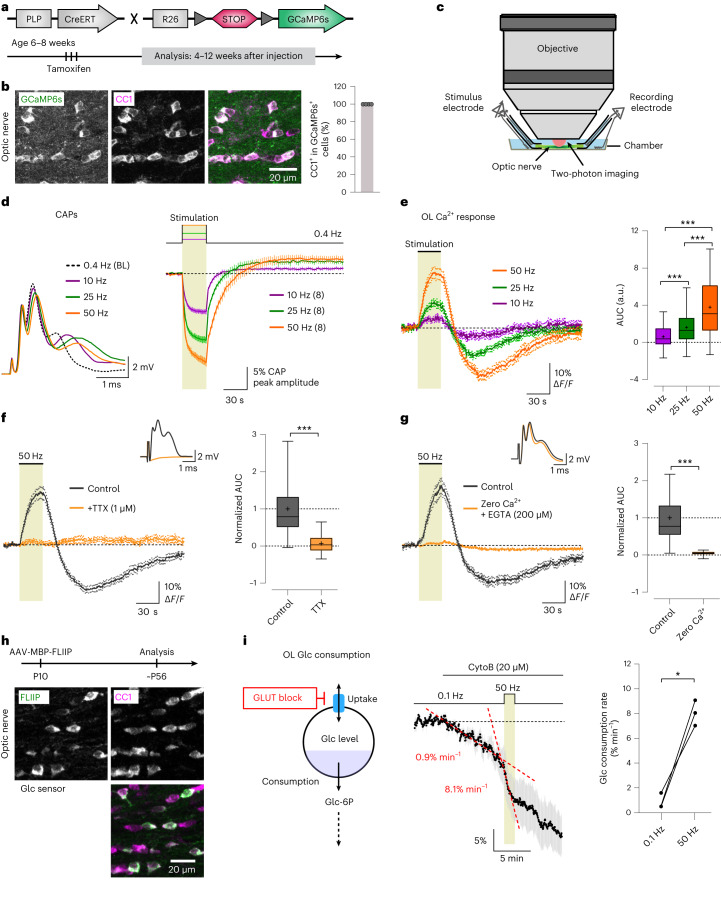
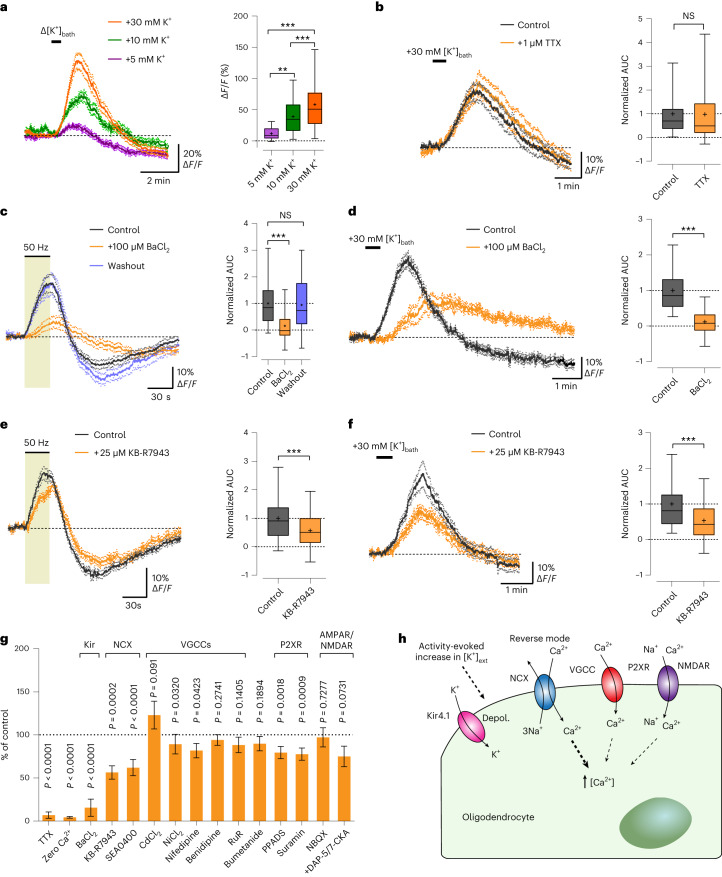
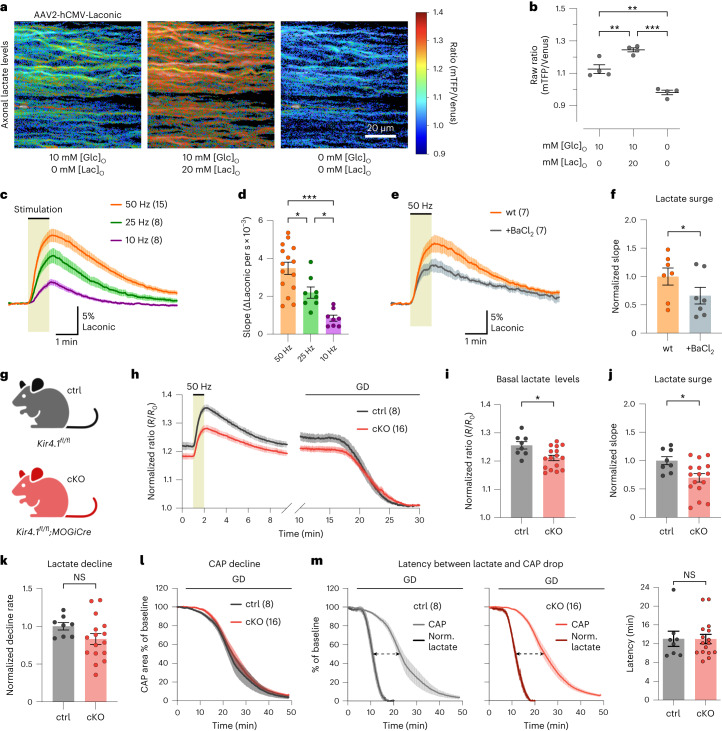
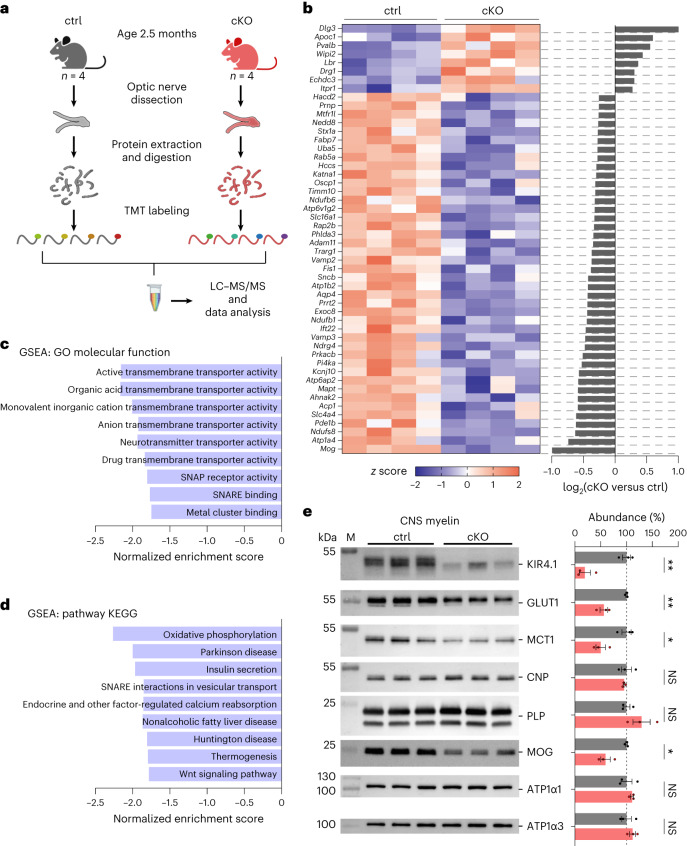
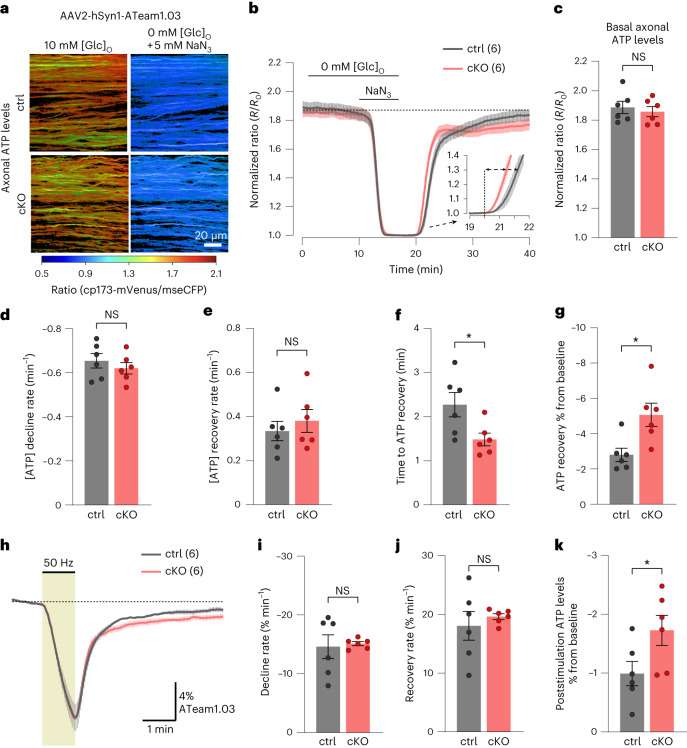
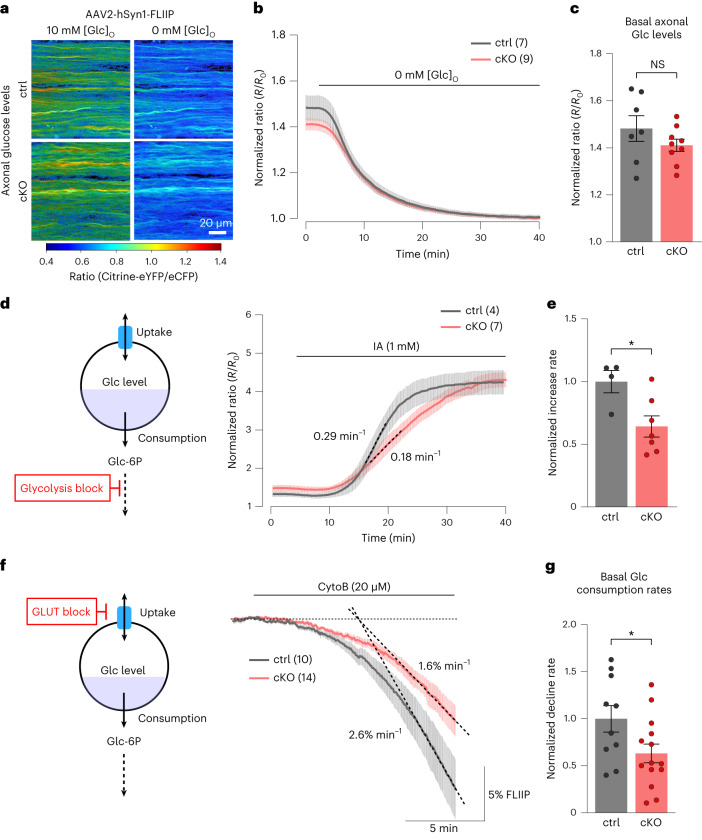
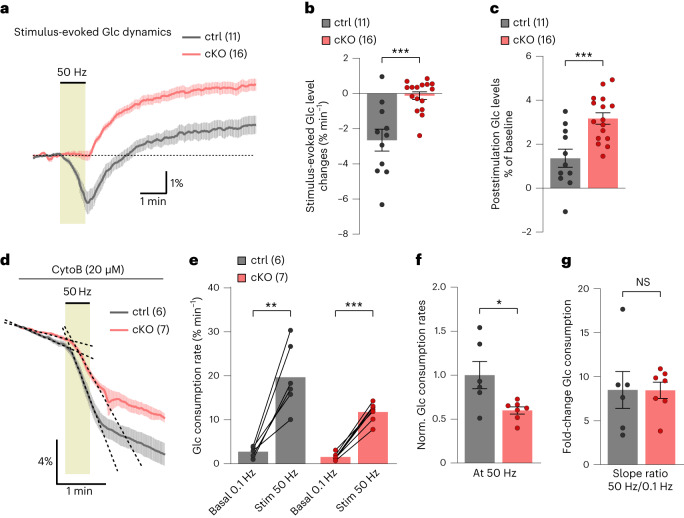
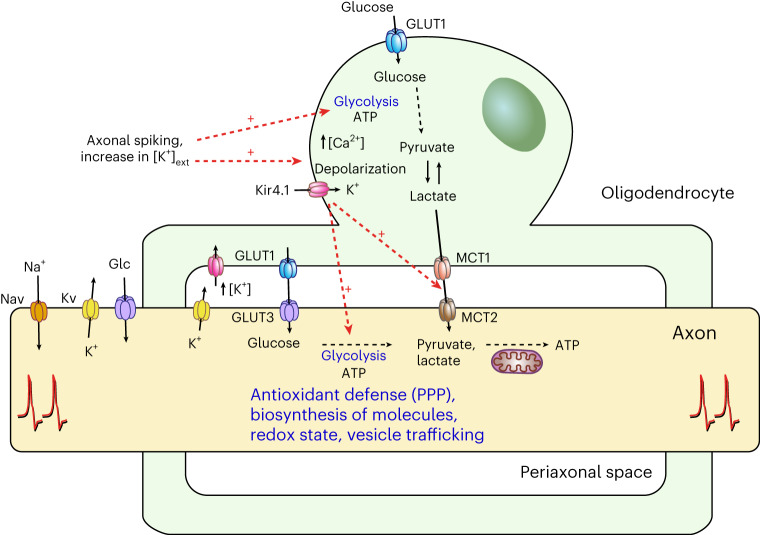
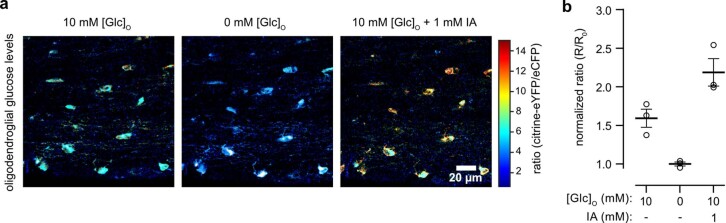
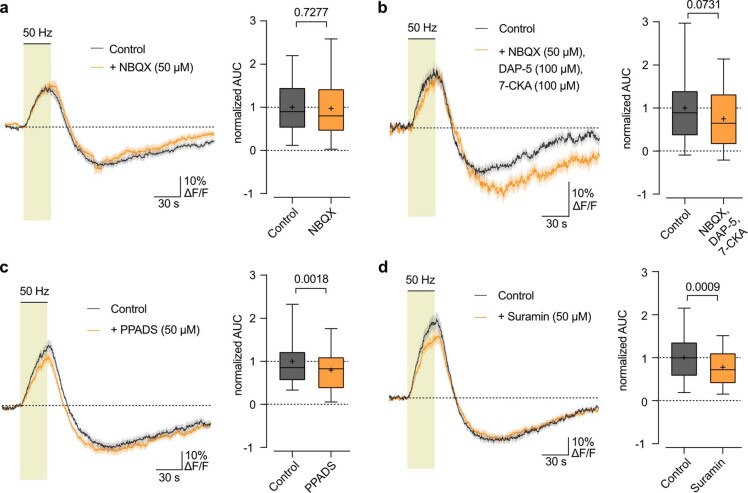
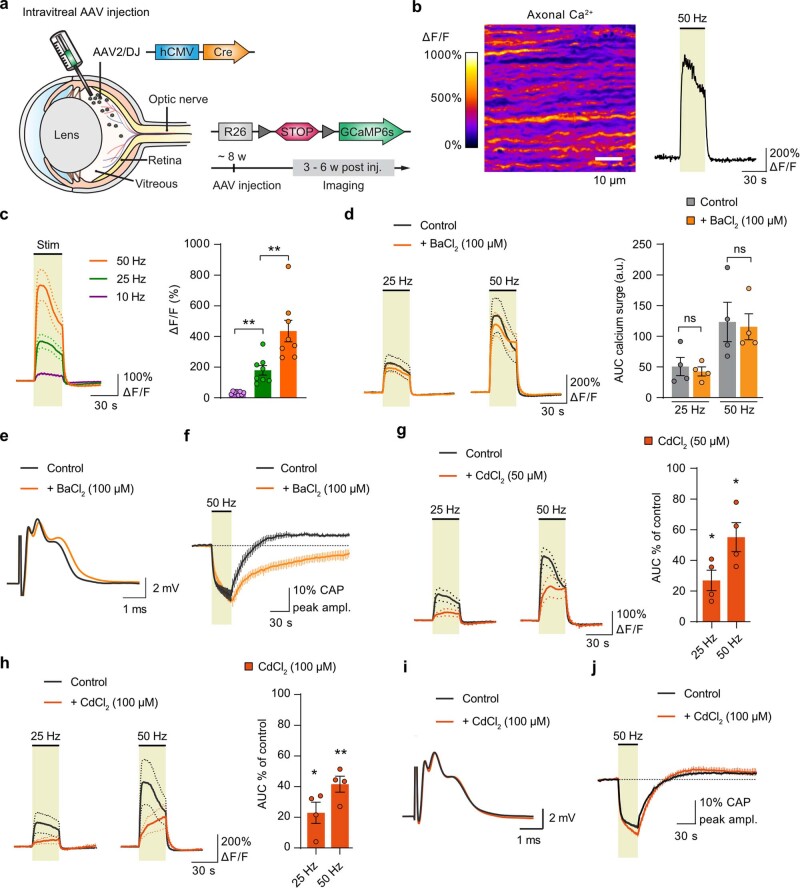
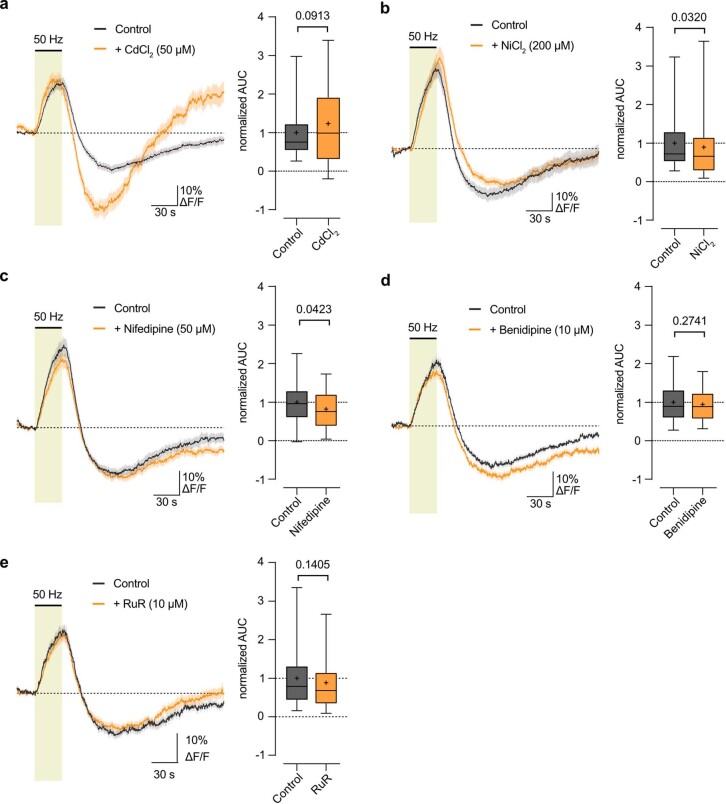
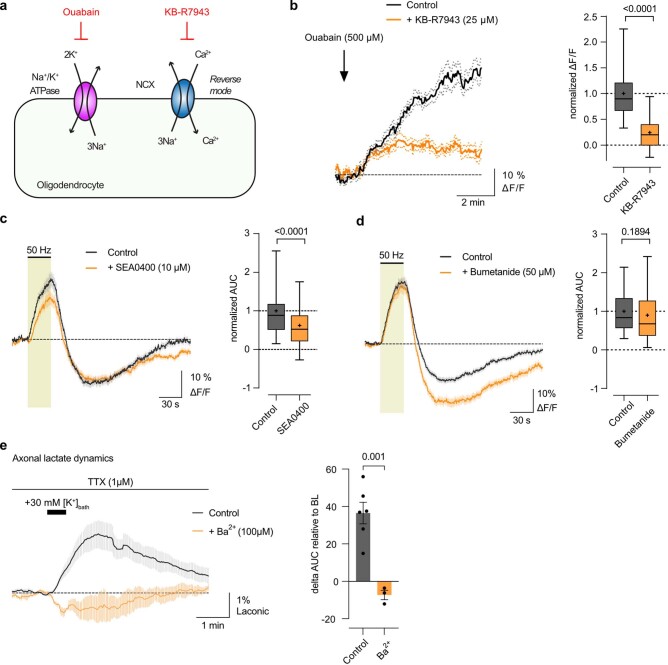
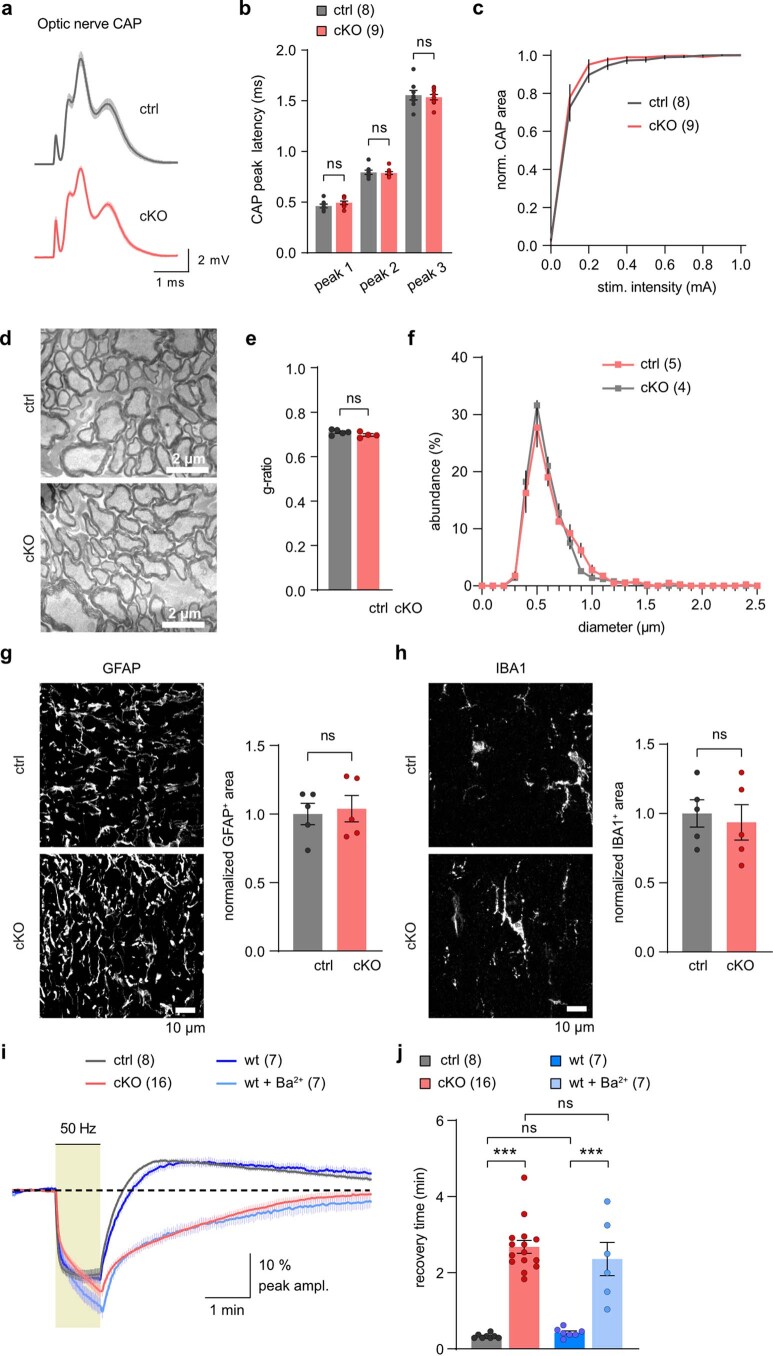
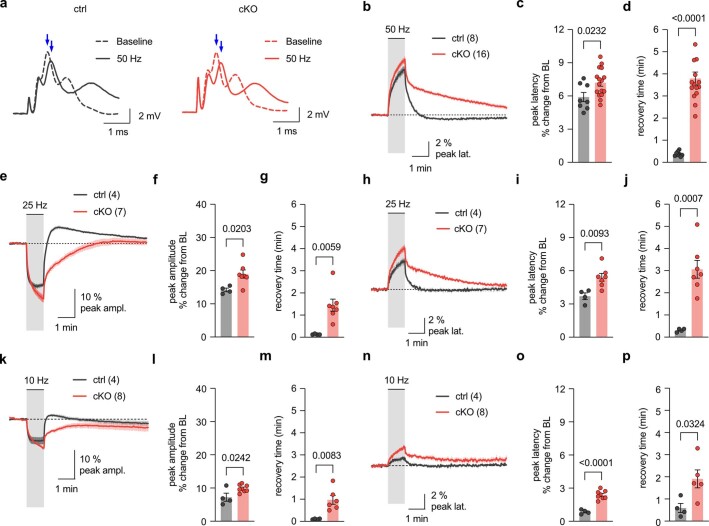
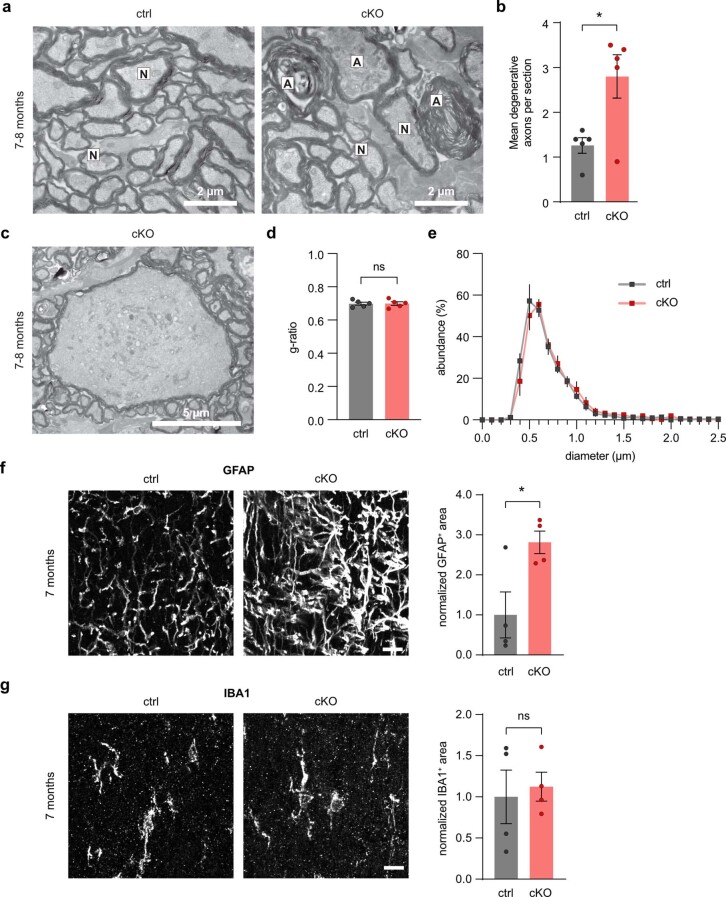
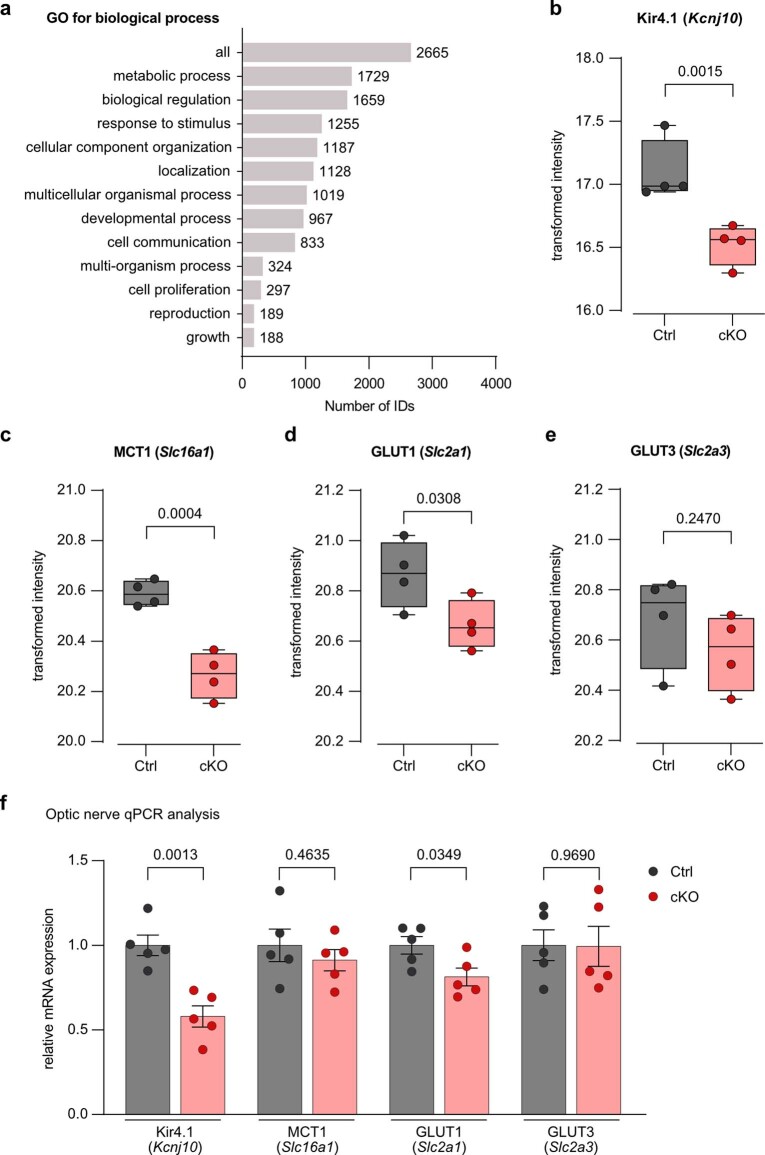
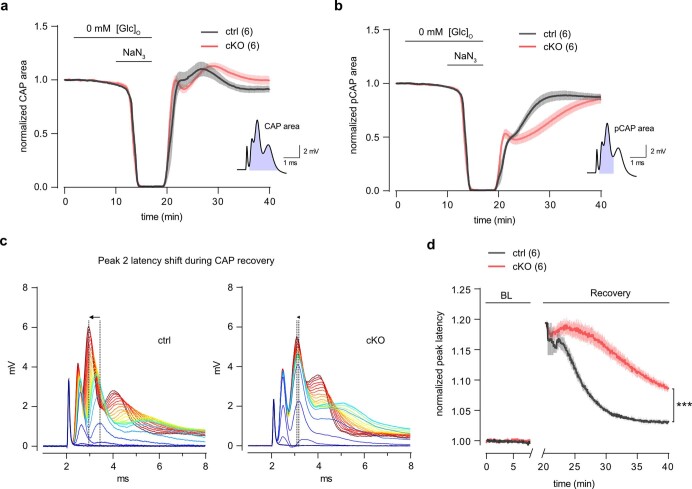
The integrity of myelinated axons relies on homeostatic support from oligodendrocytes (OLs). To determine how OLs detect axonal spiking and how rapid axon-OL metabolic coupling is regulated in the white matter, we studied activity-dependent calcium (Ca2+) and metabolite fluxes in the mouse optic nerve. We show that fast axonal spiking triggers Ca2+ signaling and glycolysis in OLs. OLs detect axonal activity through increases in extracellular potassium (K+) concentrations and activation of Kir4.1 channels, thereby regulating metabolite supply to axons. Both pharmacological inhibition and OL-specific inactivation of Kir4.1 reduce the activity-induced axonal lactate surge. Mice lacking oligodendroglial Kir4.1 exhibit lower resting lactate levels and altered glucose metabolism in axons. These early deficits in axonal energy metabolism are associated with late-onset axonopathy. Our findings reveal that OLs detect fast axonal spiking through K+ signaling, making acute metabolic coupling possible and adjusting the axon-OL metabolic unit to promote axonal health.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity.Elife. 2018 Sep 11;7:e36428. doi: 10.7554/eLife.36428. Elife. 2018. PMID: 30204081 Free PMC article.
-
Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility.Elife. 2018 Mar 29;7:e34829. doi: 10.7554/eLife.34829. Elife. 2018. PMID: 29596047 Free PMC article.
-
Can lactate serve as an energy substrate for axons in good times and in bad, in sickness and in health?Metab Brain Dis. 2015 Feb;30(1):25-30. doi: 10.1007/s11011-014-9595-3. Epub 2014 Jul 19. Metab Brain Dis. 2015. PMID: 25034458 Free PMC article. Review.
-
Oligodendrocyte-derived transcellular signaling regulates axonal energy metabolism.Curr Opin Neurobiol. 2023 Jun;80:102722. doi: 10.1016/j.conb.2023.102722. Epub 2023 Apr 5. Curr Opin Neurobiol. 2023. PMID: 37028201 Free PMC article. Review.
-
MCT1 Deletion in Oligodendrocyte Lineage Cells Causes Late-Onset Hypomyelination and Axonal Degeneration.Cell Rep. 2021 Jan 12;34(2):108610. doi: 10.1016/j.celrep.2020.108610. Cell Rep. 2021. PMID: 33440165 Free PMC article.
Cited by
-
Low intensity repetitive transcranial magnetic stimulation enhances remyelination by newborn and surviving oligodendrocytes in the cuprizone model of toxic demyelination.Cell Mol Life Sci. 2024 Aug 12;81(1):346. doi: 10.1007/s00018-024-05391-0. Cell Mol Life Sci. 2024. PMID: 39134808 Free PMC article.
-
Glycolytic dysregulation in Alzheimer's disease: unveiling new avenues for understanding pathogenesis and improving therapy.Neural Regen Res. 2025 Aug 1;20(8):2264-2278. doi: 10.4103/NRR.NRR-D-24-00190. Epub 2024 Jul 29. Neural Regen Res. 2025. PMID: 39101629 Free PMC article.
-
Oligodendrocytes in Alzheimer's disease pathophysiology.Nat Neurosci. 2025 Mar;28(3):446-456. doi: 10.1038/s41593-025-01873-x. Epub 2025 Jan 29. Nat Neurosci. 2025. PMID: 39881195 Review.
-
Developmental maturation and regional heterogeneity but no sexual dimorphism of the murine CNS myelin proteome.Glia. 2025 Jan;73(1):38-56. doi: 10.1002/glia.24614. Epub 2024 Sep 30. Glia. 2025. PMID: 39344832 Free PMC article.
-
An Overview of Oligodendrocyte Metabolism.Adv Neurobiol. 2025;43:155-179. doi: 10.1007/978-3-031-87919-7_7. Adv Neurobiol. 2025. PMID: 40500498 Review.
References
-
- Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. - PubMed
-
- Saab AS, Tzvetanova ID, Nave K-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013;23:1065–1072. - PubMed
-
- Nave K-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010;11:275–283. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
