

In vitro characterization of rare anti-αIIbβ3 isoantibodies produced by patients with Glanzmann thrombasthenia that severely block fibrinogen binding and generate procoagulant platelets via complement activation
- PMID: 38268518
- PMCID: PMC10805943
- DOI: 10.1016/j.rpth.2023.102253
In vitro characterization of rare anti-αIIbβ3 isoantibodies produced by patients with Glanzmann thrombasthenia that severely block fibrinogen binding and generate procoagulant platelets via complement activation
Abstract
Background: Glanzmann thrombasthenia (GT) is a rare bleeding disorder caused by inherited defects of the platelet αIIbβ3 integrin. Platelet transfusions can be followed by an immune response that can block integrin function by interfering with fibrinogen binding.
Objectives: In this study, we aimed to determine the prevalence of such isoantibodies and better characterize their pathogenic properties.
Methods: Twelve patients with GT were evaluated for anti-αIIbβ3 isoantibodies. Sera from patients with GT with or without anti-αIIbβ3 isoantibodies were then used to study their in vitro effect on platelets from healthy donors. We used several approaches (IgG purification, immunofluorescence staining, and inhibition of signaling pathways) to characterize the pathogenic properties of the anti-αIIbβ3 isoantibodies.
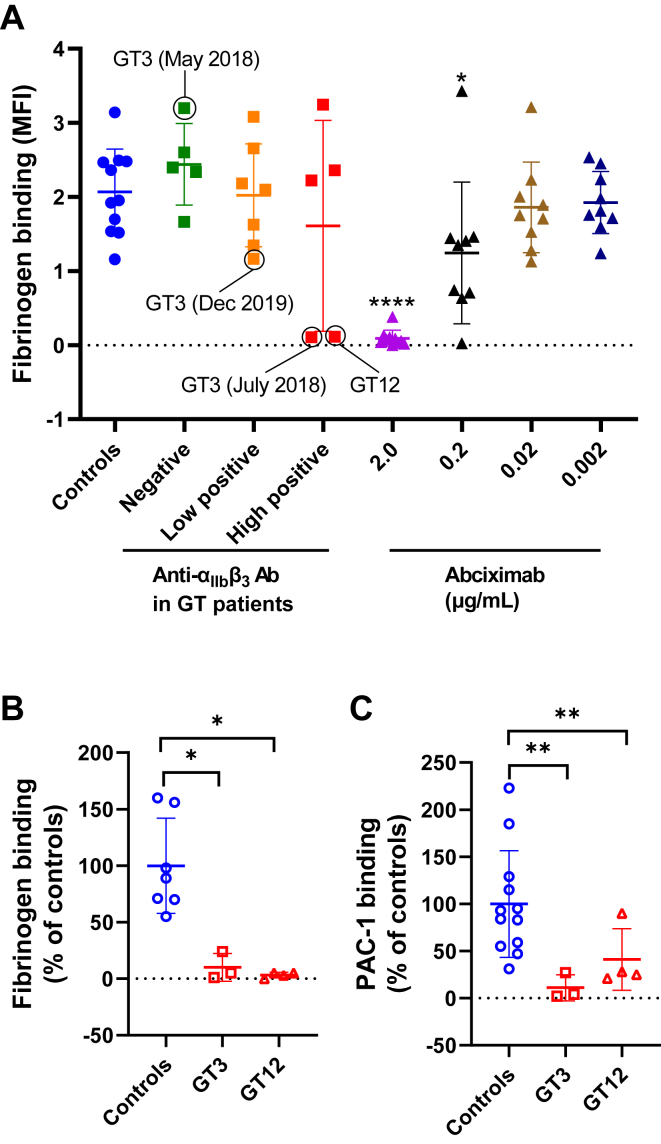
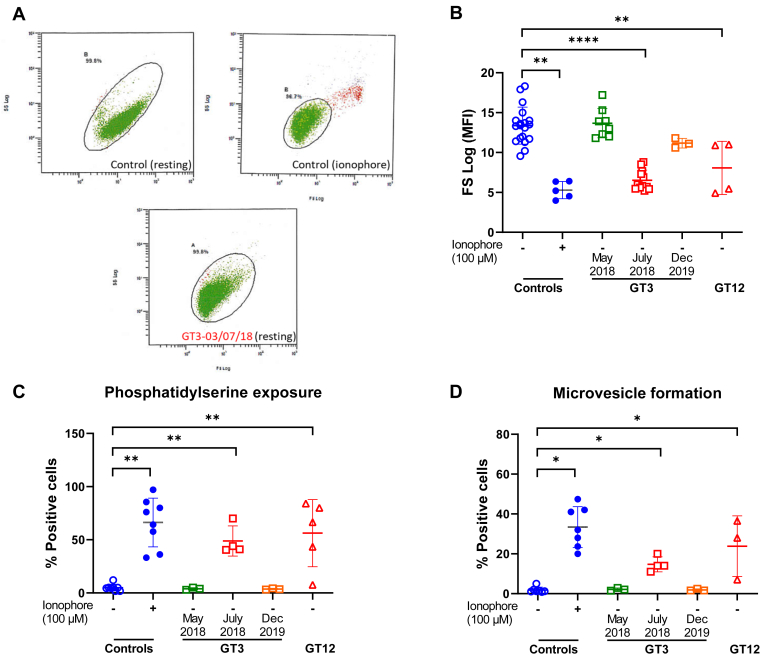
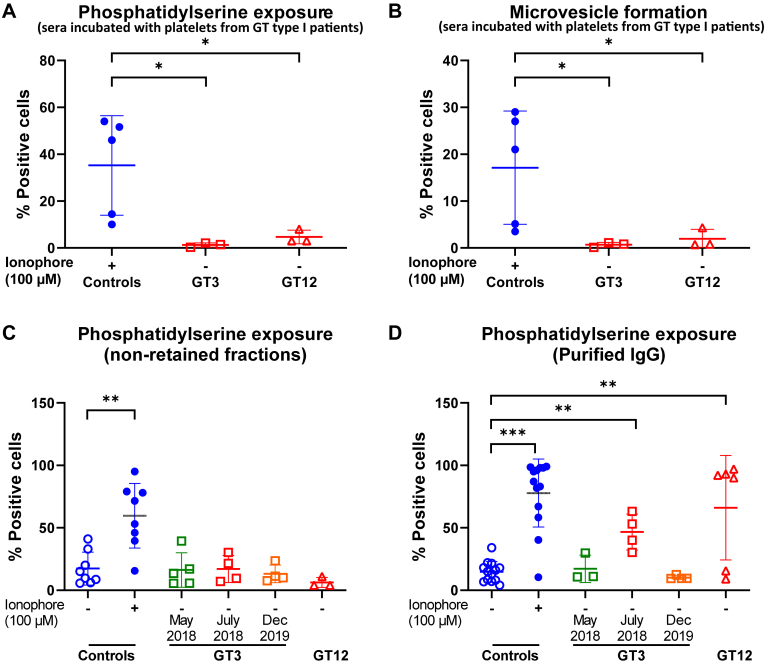
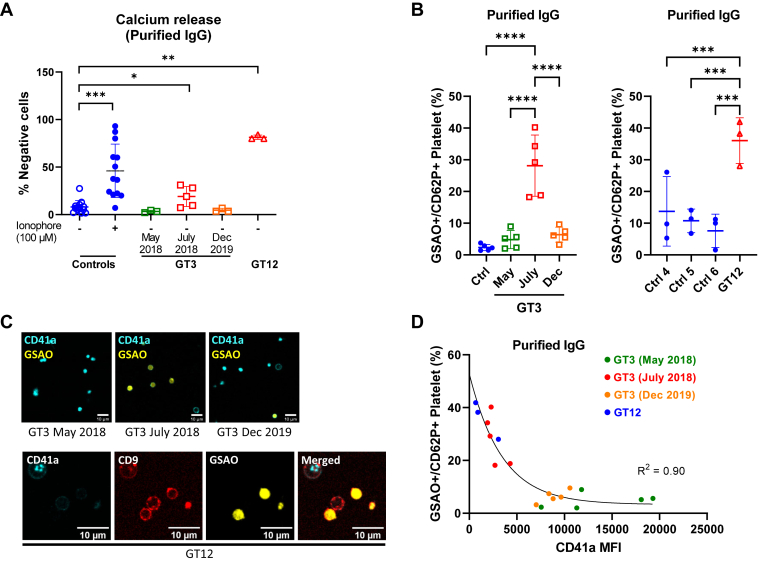
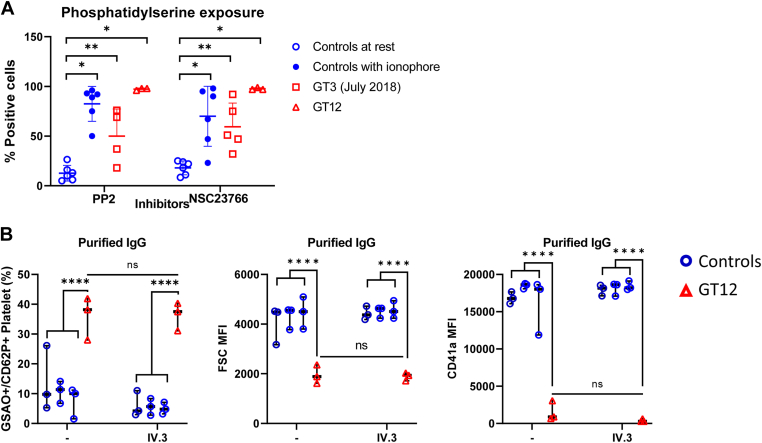
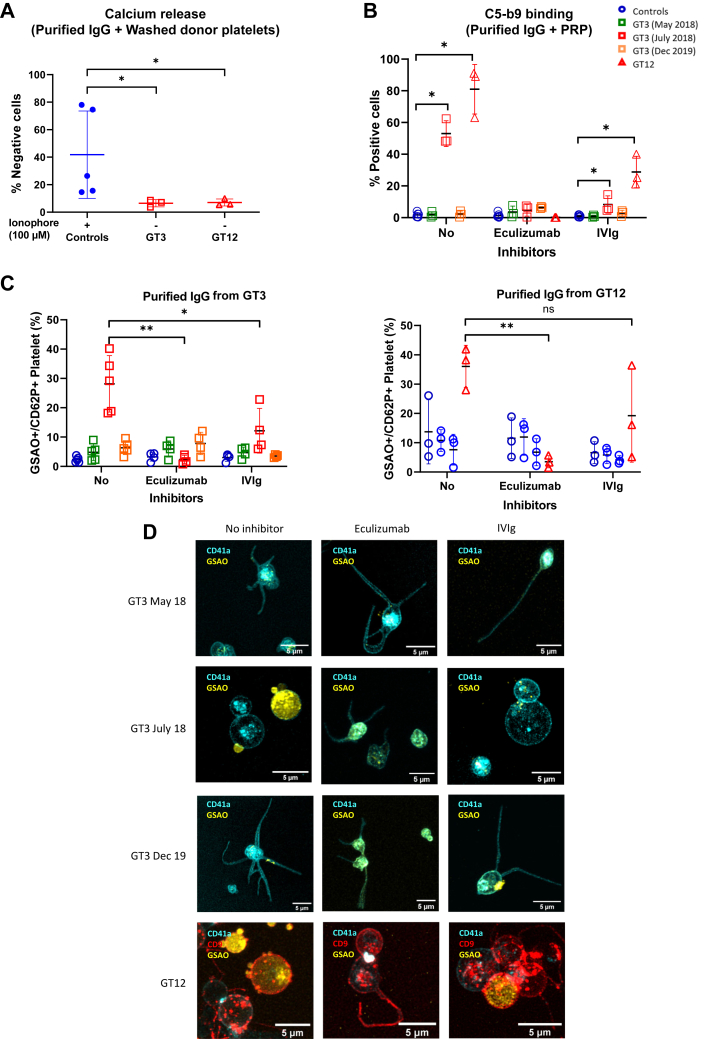
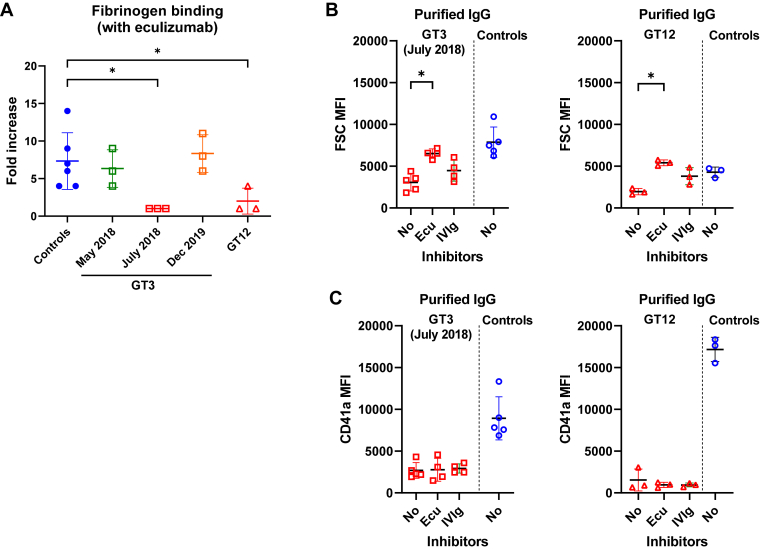
Results: Only 2 samples were able to severely block integrin function. We observed that these 2 sera caused a reduction in platelet size similar to that observed when platelets become procoagulant. Mixing healthy donor platelets with patients' sera or purified IgGs led to microvesiculation, phosphatidylserine exposure, and induction of calcium influx. This was associated with an increase in procoagulant platelets. Pore formation and calcium entry were associated with complement activation, leading to the constitution of a membrane attack complex (MAC) with enhanced complement protein C5b-9 formation. This process was inhibited by the complement 5 inhibitor eculizumab and reduced by polyvalent human immunoglobulins.
Conclusion: Our data suggest that complement activation induced by rare blocking anti-αIIbβ3 isoantibodies may lead to the formation of a MAC with subsequent pore formation, resulting in calcium influx and procoagulant platelet phenotype.
Keywords: Glanzmann thrombasthenia; anti-αIIbβ3 isoantibodies; coagulation; complement activation; platelet transfusion; procoagulant platelet.
© 2023 The Authors.
Figures
References
-
- Nurden A.T. Platelet membrane glycoproteins: a historical review. Semin Thromb Hemost. 2014;40:577–584. - PubMed
-
- Nurden A.T., Fiore M., Nurden P., Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
