

Characterizing adjuvants' effects at murine immunoglobulin repertoire level
- PMID: 38269092
- PMCID: PMC10805652
- DOI: 10.1016/j.isci.2023.108749
Characterizing adjuvants' effects at murine immunoglobulin repertoire level
Abstract
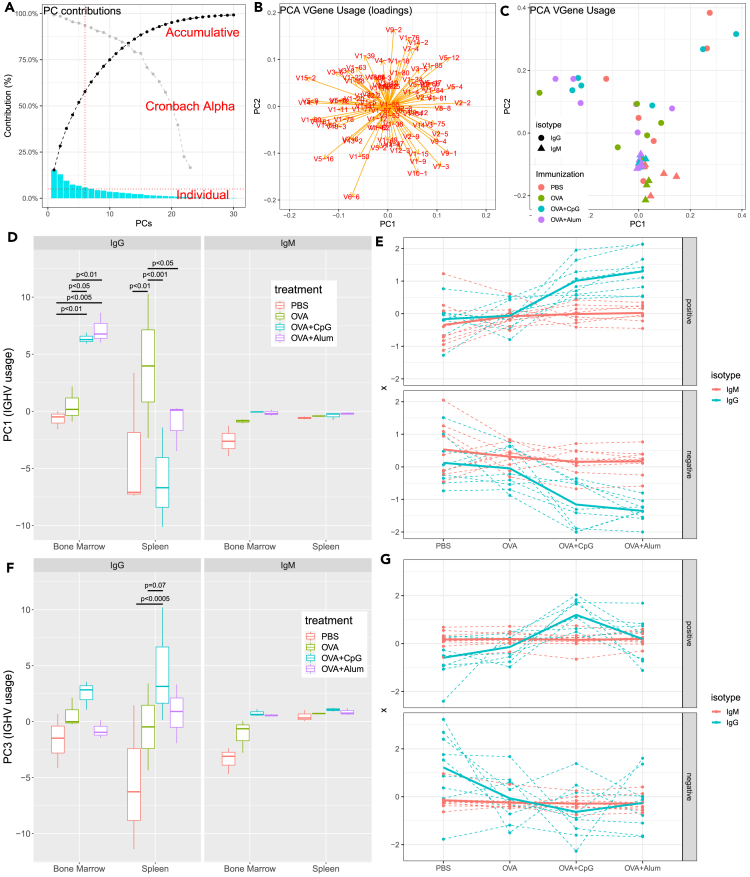
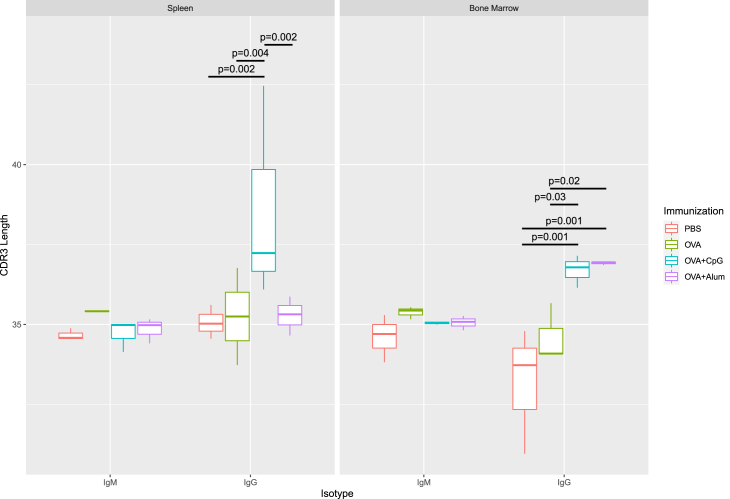
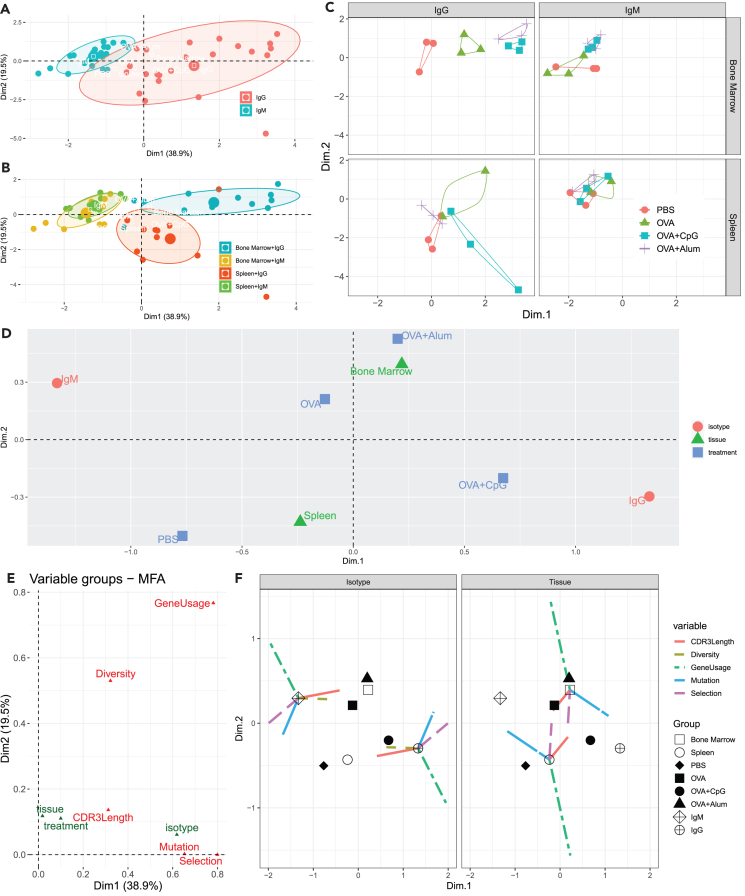
Generating large-scale, high-fidelity sequencing data is challenging and, furthermore, not much has been done to characterize adjuvants' effects at the repertoire level. Thus, we introduced an IgSeq pipeline that standardized library prep protocols and data analysis functions for accurate repertoire profiling. We then studied systemically effects of CpG and Alum on the Ig heavy chain repertoire using the ovalbumin (OVA) murine model. Ig repertoires of different tissues (spleen and bone marrow) and isotypes (IgG and IgM) were examined and compared in IGHV mutation, gene usage, CDR3 length, clonal diversity, and clonal selection. We found Ig repertoires of different compartments exhibited distinguishable profiles at the non-immunized steady state, and distinctions became more pronounced upon adjuvanted immunizations. Notably, Alum and CpG effects exhibited different tissue- and isotype-preferences. The former led to increased diversity of abundant clones in bone marrow, and the latter promoted the selection of IgG clones in both tissues.
Keywords: Genomic analysis; Immunology; Machine learning; Sequence analysis.
© 2023.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures





References
-
- Galson J.D., Trück J., Fowler A., Clutterbuck E.A., Münz M., Cerundolo V., Reinhard C., van der Most R., Pollard A.J., Lunter G., Kelly D.F. Analysis of B Cell Repertoire Dynamics Following Hepatitis B Vaccination in Humans, and Enrichment of Vaccine-specific Antibody Sequences. EBioMedicine. 2015;2:2070–2079. - PMC - PubMed
-
- Galson J.D., Schaetzle S., Bashford-Rogers R.J.M., Raybould M.I.J., Kovaltsuk A., Kilpatrick G.J., Minter R., Finch D.K., Dias J., James L.K., et al. Deep sequencing of b cell receptor repertoires from covid-19 patients reveals strong convergent immune signatures. Front. Immunol. 2020;11 - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
