

Reconstructing growth and dynamic trajectories from single-cell transcriptomics data
- PMID: 38274364
- PMCID: PMC10805654
- DOI: 10.1038/s42256-023-00763-w
Reconstructing growth and dynamic trajectories from single-cell transcriptomics data
Abstract
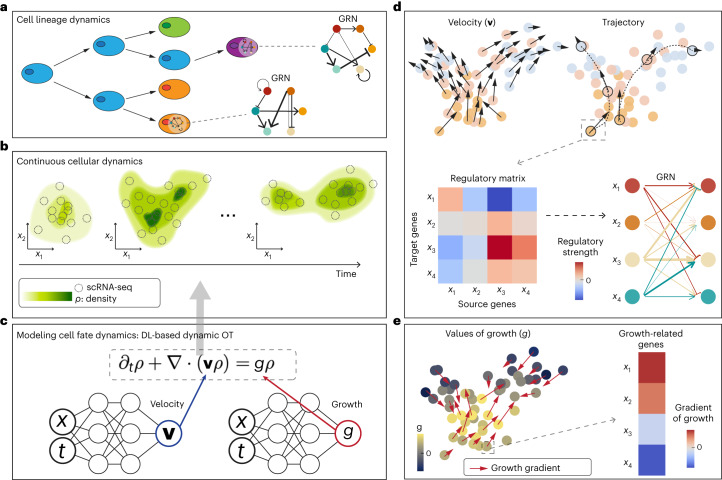
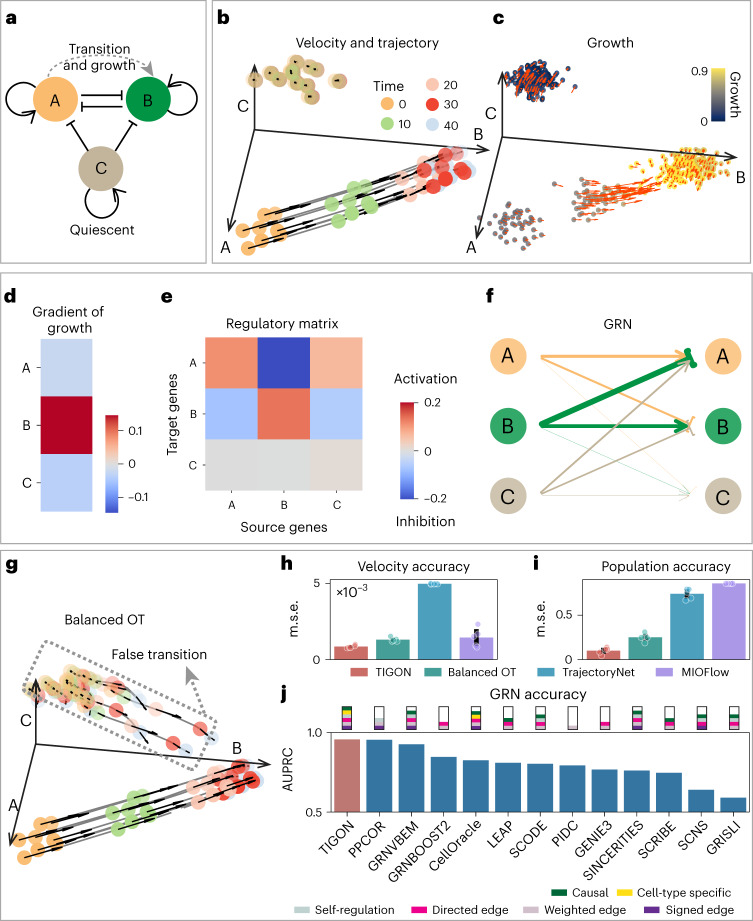
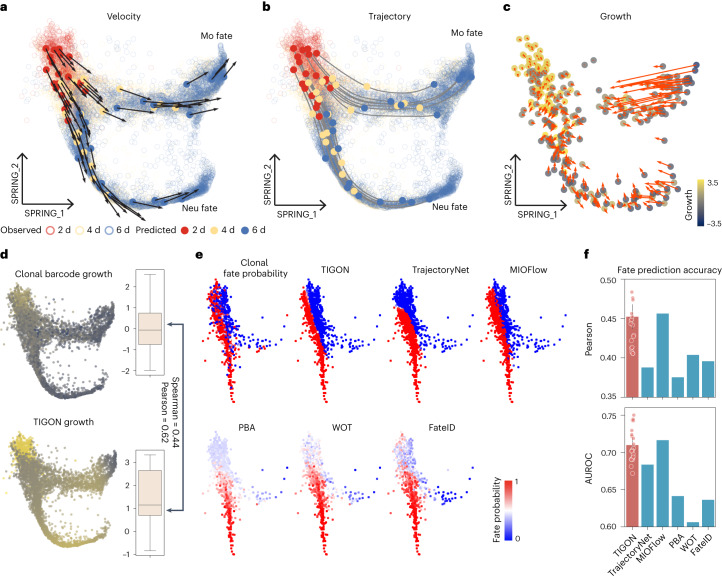
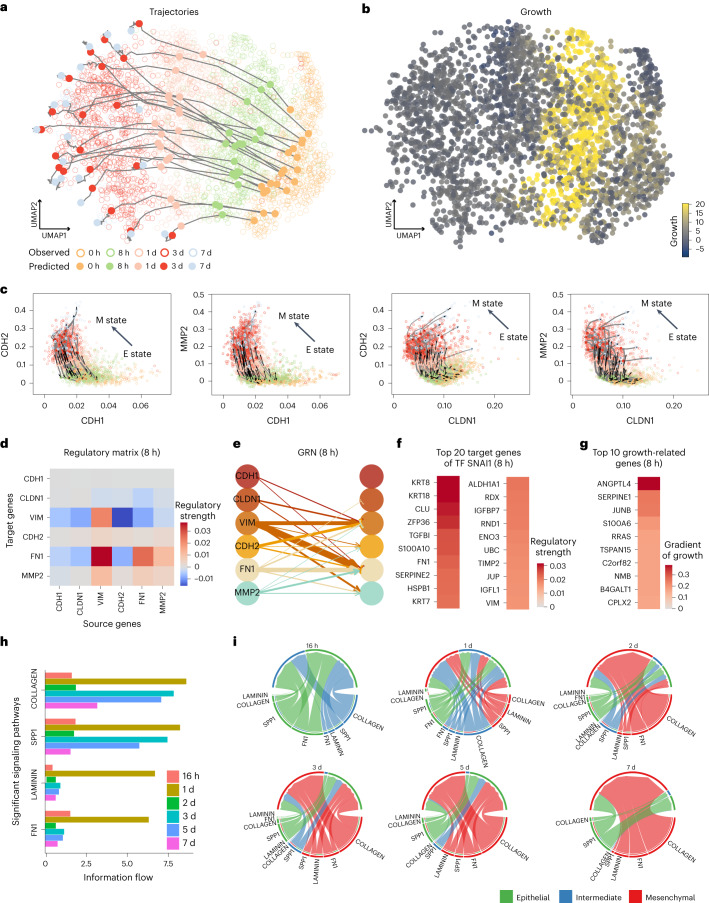
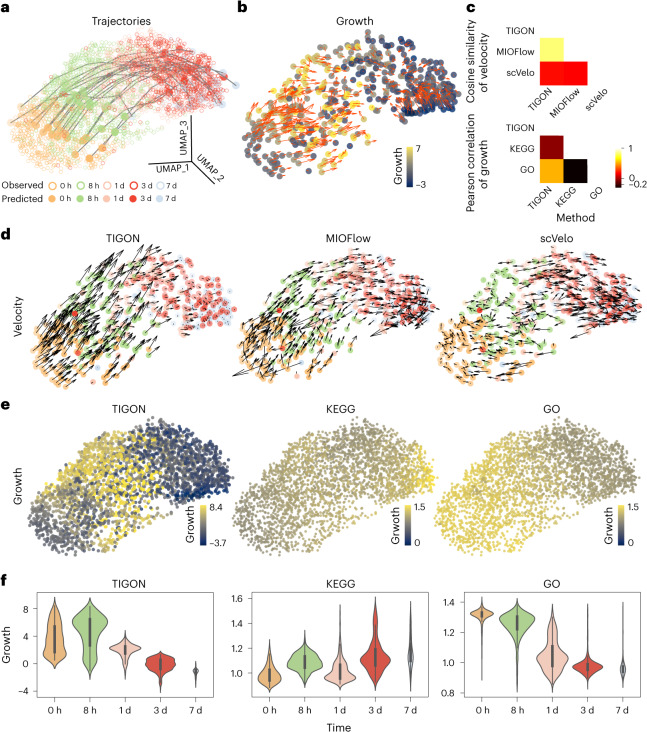
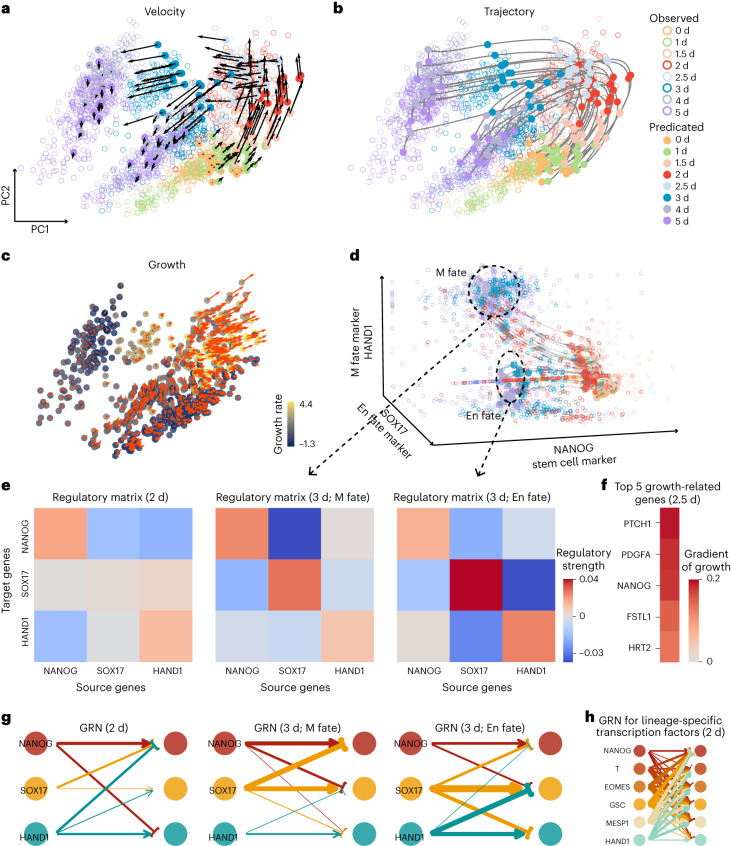
Time-series single-cell RNA sequencing (scRNA-seq) datasets provide unprecedented opportunities to learn dynamic processes of cellular systems. Due to the destructive nature of sequencing, it remains challenging to link the scRNA-seq snapshots sampled at different time points. Here we present TIGON, a dynamic, unbalanced optimal transport algorithm that reconstructs dynamic trajectories and population growth simultaneously as well as the underlying gene regulatory network from multiple snapshots. To tackle the high-dimensional optimal transport problem, we introduce a deep learning method using a dimensionless formulation based on the Wasserstein-Fisher-Rao (WFR) distance. TIGON is evaluated on simulated data and compared with existing methods for its robustness and accuracy in predicting cell state transition and cell population growth. Using three scRNA-seq datasets, we show the importance of growth in the temporal inference, TIGON's capability in reconstructing gene expression at unmeasured time points and its applications to temporal gene regulatory networks and cell-cell communication inference.
Keywords: Data integration; Machine learning.
© The Author(s) 2023, corrected publication 2023.
Conflict of interest statement
Competing interestsThe authors declare no competing interests.
Figures
Similar articles
-
Predicting gene regulatory links from single-cell RNA-seq data using graph neural networks.Brief Bioinform. 2023 Sep 22;24(6):bbad414. doi: 10.1093/bib/bbad414. Brief Bioinform. 2023. PMID: 37985457 Free PMC article.
-
Inferring Gene Regulatory Networks From Single-Cell Transcriptomic Data Using Bidirectional RNN.Front Oncol. 2022 May 26;12:899825. doi: 10.3389/fonc.2022.899825. eCollection 2022. Front Oncol. 2022. PMID: 35692809 Free PMC article.
-
Dynamic inference of cell developmental complex energy landscape from time series single-cell transcriptomic data.PLoS Comput Biol. 2022 Jan 24;18(1):e1009821. doi: 10.1371/journal.pcbi.1009821. eCollection 2022 Jan. PLoS Comput Biol. 2022. PMID: 35073331 Free PMC article.
-
Integrating Dynamical Systems Modeling with Spatiotemporal scRNA-Seq Data Analysis.Entropy (Basel). 2025 Apr 22;27(5):453. doi: 10.3390/e27050453. Entropy (Basel). 2025. PMID: 40422408 Free PMC article. Review.
-
A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell-Cell Communication.Cells. 2023 Jul 30;12(15):1970. doi: 10.3390/cells12151970. Cells. 2023. PMID: 37566049 Free PMC article. Review.
Cited by
-
Reconstruction of single-cell lineage trajectories and identification of diversity in fates during the epithelial-to-mesenchymal transition.Proc Natl Acad Sci U S A. 2024 Aug 6;121(32):e2406842121. doi: 10.1073/pnas.2406842121. Epub 2024 Aug 2. Proc Natl Acad Sci U S A. 2024. PMID: 39093947 Free PMC article.
-
Data- and theory-driven approaches for understanding paths of epithelial-mesenchymal transition.Genesis. 2024 Apr;62(2):e23591. doi: 10.1002/dvg.23591. Genesis. 2024. PMID: 38553870 Free PMC article. Review.
-
Variational inference of single cell time series.bioRxiv [Preprint]. 2025 May 16:2024.08.29.610389. doi: 10.1101/2024.08.29.610389. bioRxiv. 2025. PMID: 39257806 Free PMC article. Preprint.
-
Reconstructing Waddington Landscape from Cell Migration and Proliferation.Interdiscip Sci. 2025 Sep;17(3):541-554. doi: 10.1007/s12539-024-00686-z. Epub 2025 Jan 7. Interdiscip Sci. 2025. PMID: 39775538
-
Optimal transport reveals dynamic gene regulatory networks via gene velocity estimation.bioRxiv [Preprint]. 2024 Sep 16:2024.09.12.612590. doi: 10.1101/2024.09.12.612590. bioRxiv. 2024. Update in: PLoS Comput Biol. 2025 May 8;21(5):e1012476. doi: 10.1371/journal.pcbi.1012476. PMID: 39345416 Free PMC article. Updated. Preprint.