

Protein Kinase A Is a Master Regulator of Physiological and Pathological Cardiac Hypertrophy
- PMID: 38275112
- PMCID: PMC10923071
- DOI: 10.1161/CIRCRESAHA.123.322729
Protein Kinase A Is a Master Regulator of Physiological and Pathological Cardiac Hypertrophy
Abstract
Background: The sympathoadrenergic system and its major effector PKA (protein kinase A) are activated to maintain cardiac output coping with physiological or pathological stressors. If and how PKA plays a role in physiological cardiac hypertrophy (PhCH) and pathological CH (PaCH) are not clear.
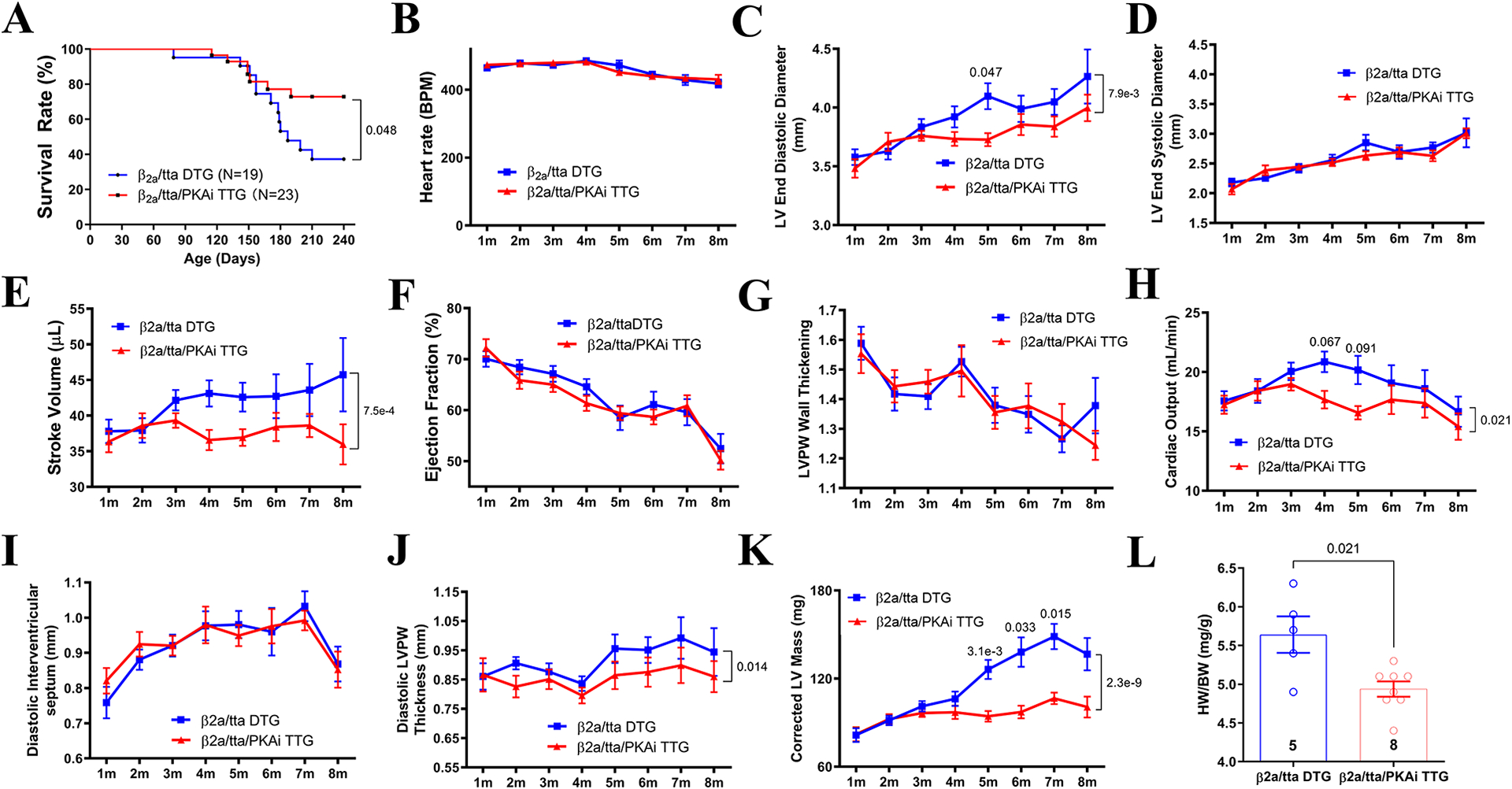
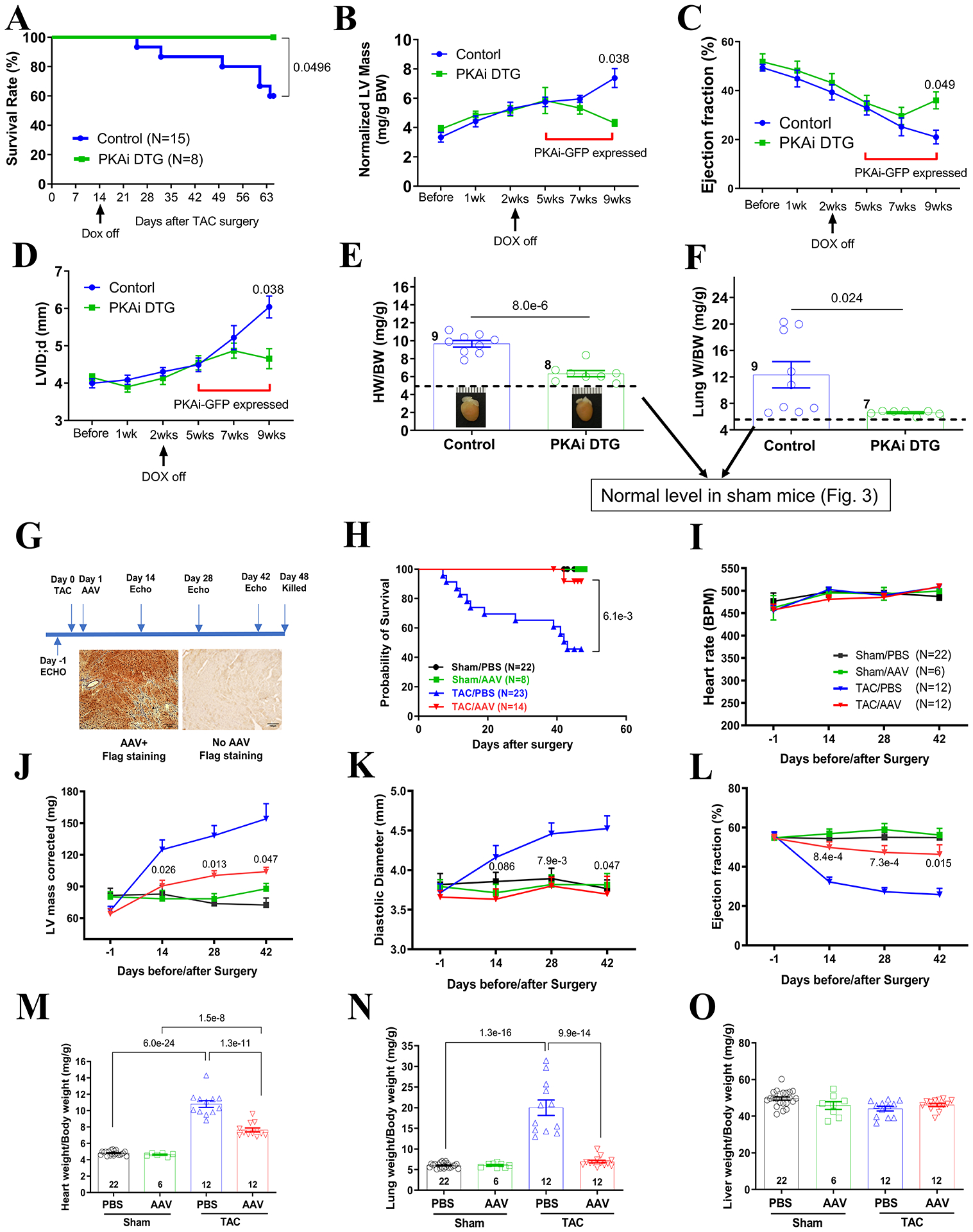
Methods: Transgenic mouse models expressing the PKA inhibition domain (PKAi) of PKA inhibition peptide alpha (PKIalpha)-green fluorescence protein (GFP) fusion protein (PKAi-GFP) in a cardiac-specific and inducible manner (cPKAi) were used to determine the roles of PKA in physiological CH during postnatal growth or induced by swimming, and in PaCH induced by transaortic constriction (TAC) or augmented Ca2+ influx. Kinase profiling was used to determine cPKAi specificity. Echocardiography was used to determine cardiac morphology and function. Western blotting and immunostaining were used to measure protein abundance and phosphorylation. Protein synthesis was assessed by puromycin incorporation and protein degradation by measuring protein ubiquitination and proteasome activity. Neonatal rat cardiomyocytes (NRCMs) infected with AdGFP (GFP adenovirus) or AdPKAi-GFP (PKAi-GFP adenovirus) were used to determine the effects and mechanisms of cPKAi on myocyte hypertrophy. rAAV9.PKAi-GFP was used to treat TAC mice.
Results: (1) cPKAi delayed postnatal cardiac growth and blunted exercise-induced PhCH; (2) PKA was activated in hearts after TAC due to activated sympathoadrenergic system, the loss of endogenous PKIα (PKA inhibition peptide α), and the stimulation by noncanonical PKA activators; (3) cPKAi ameliorated PaCH induced by TAC and increased Ca2+ influxes and blunted neonatal rat cardiomyocyte hypertrophy by isoproterenol and phenylephrine; (4) cPKAi prevented TAC-induced protein synthesis by inhibiting mTOR (mammalian target of rapamycin) signaling through reducing Akt (protein kinase B) activity, but enhancing inhibitory GSK-3α (glycogen synthase kinase-3α) and GSK-3β signals; (5) cPKAi reduced protein degradation by the ubiquitin-proteasome system via decreasing RPN6 phosphorylation; (6) cPKAi increased the expression of antihypertrophic atrial natriuretic peptide (ANP); (7) cPKAi ameliorated established PaCH and improved animal survival.
Conclusions: Cardiomyocyte PKA is a master regulator of PhCH and PaCH through regulating protein synthesis and degradation. cPKAi can be a novel approach to treat PaCH.
Keywords: cyclic AMP-dependent protein kinases; heart; hypertrophy; mechanistic target of rapamycin complex 1; proteolysis.
Conflict of interest statement
Figures






References
-
- Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407 - PubMed
-
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: A new therapeutic target? Circulation. 2004;109:1580–1589 - PubMed
-
- Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89:997–1004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
