

scPerturb: harmonized single-cell perturbation data
- PMID: 38279009
- PMCID: PMC12220817
- DOI: 10.1038/s41592-023-02144-y
scPerturb: harmonized single-cell perturbation data
Abstract
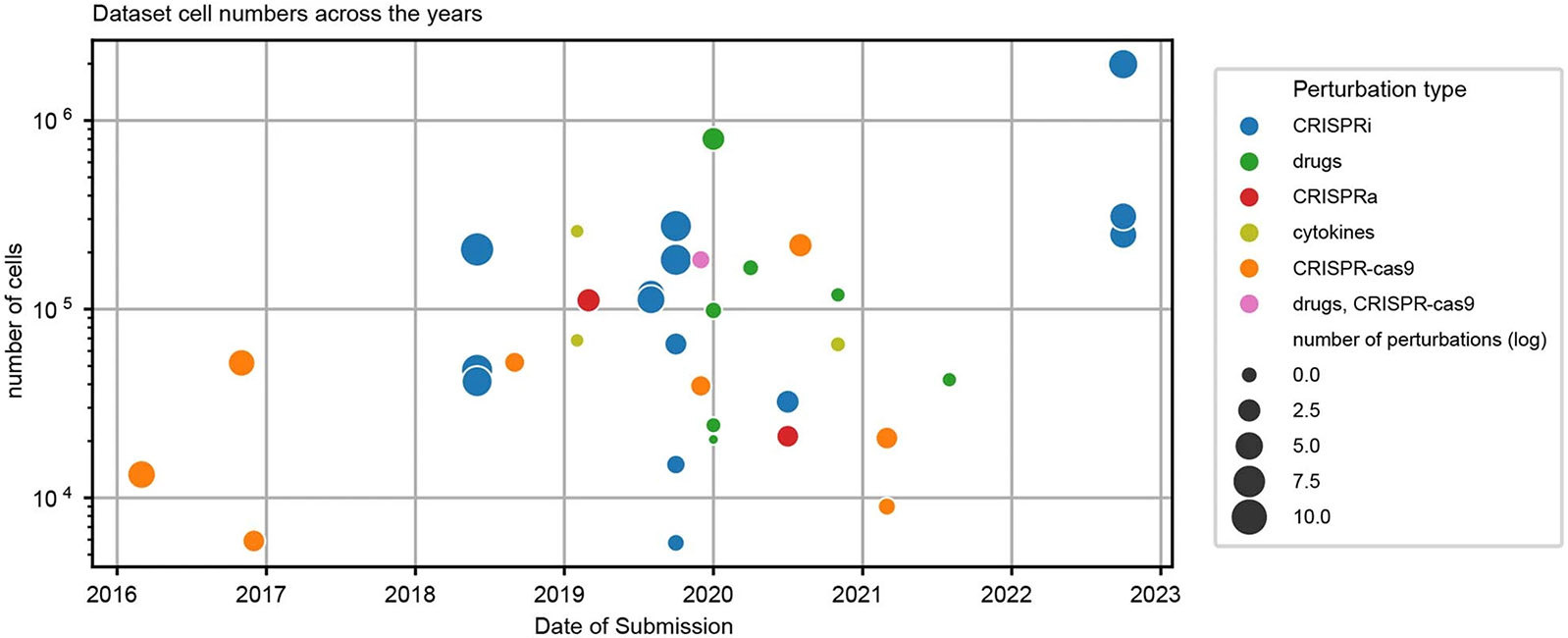
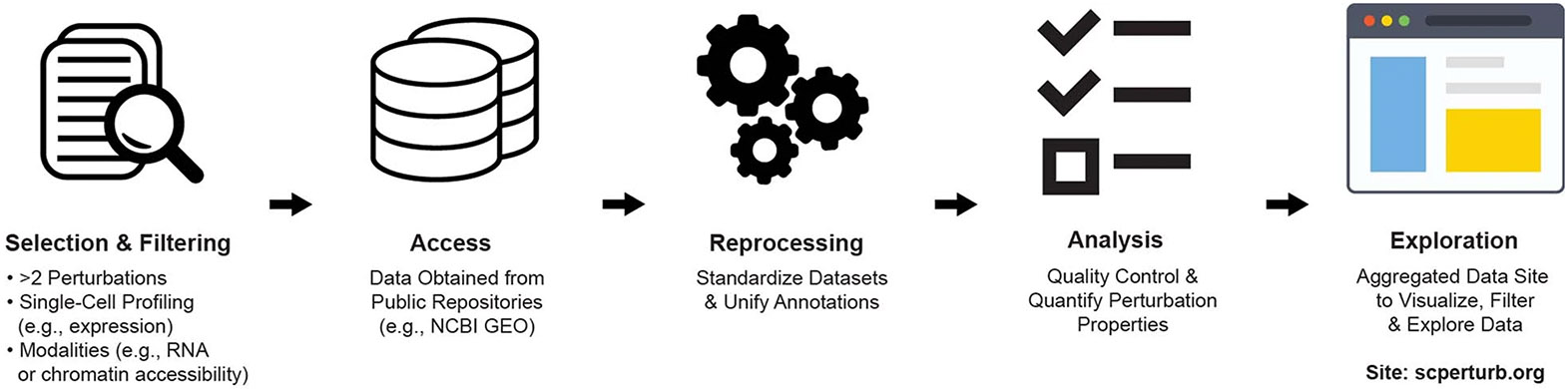
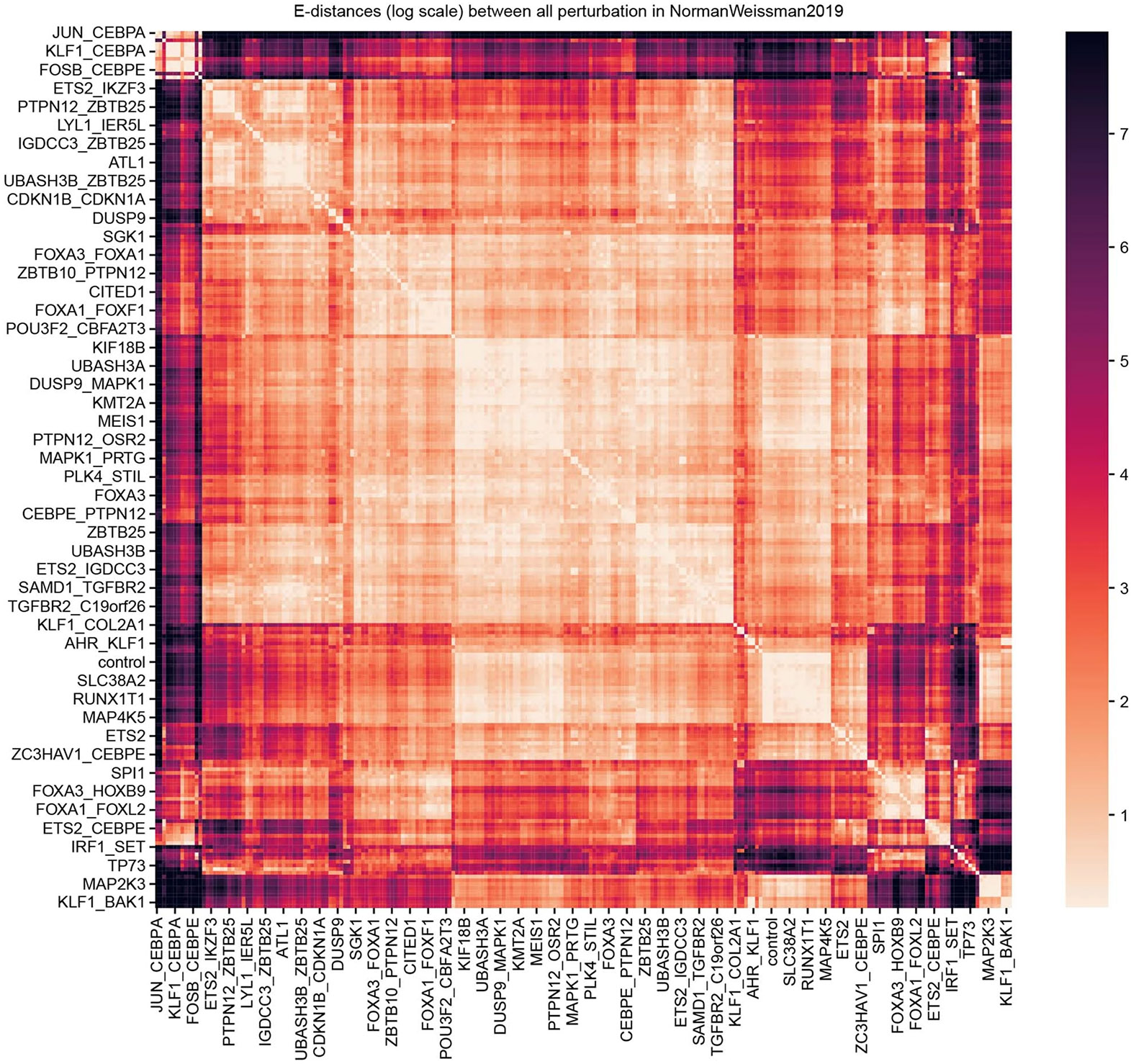
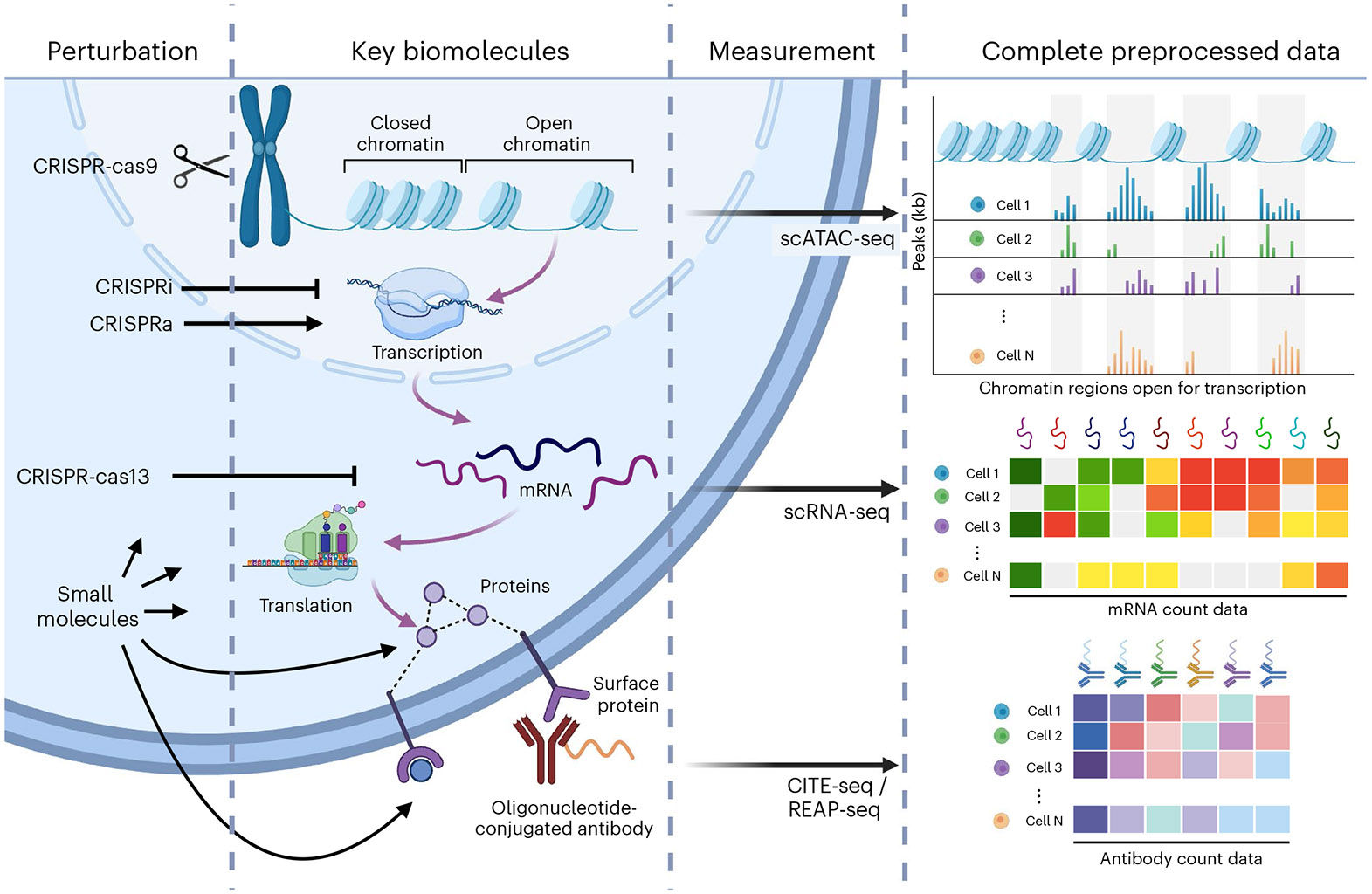
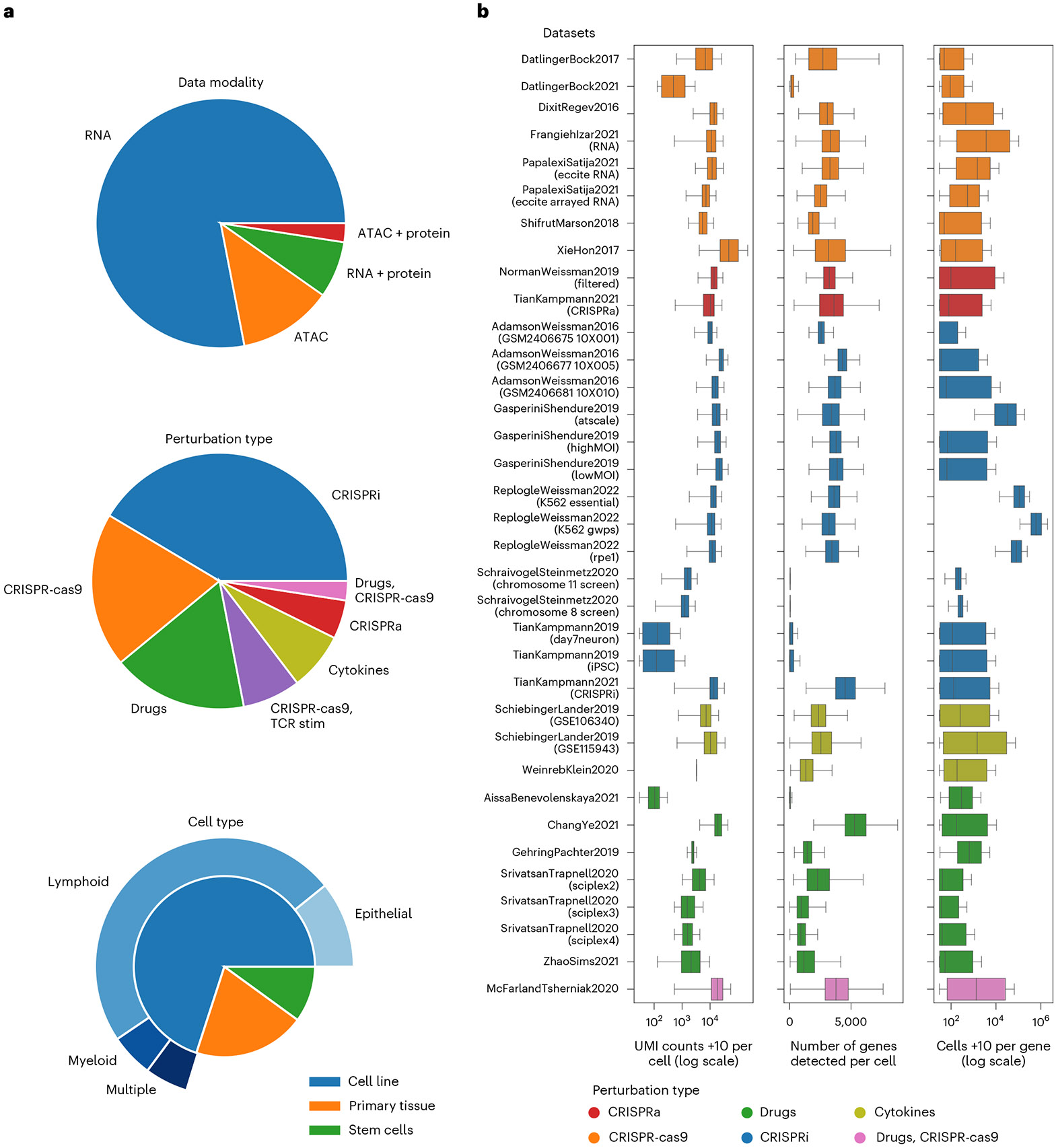
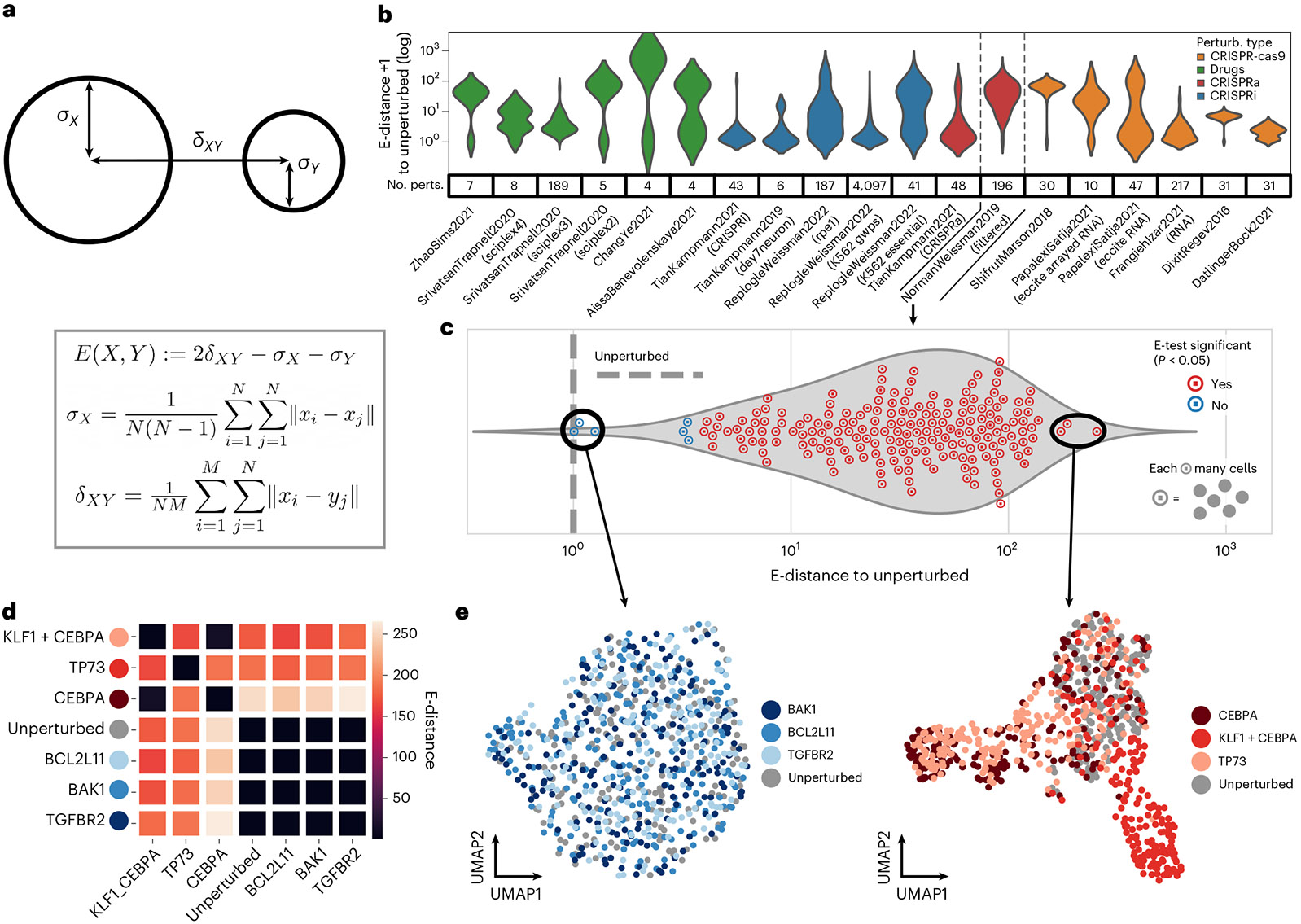
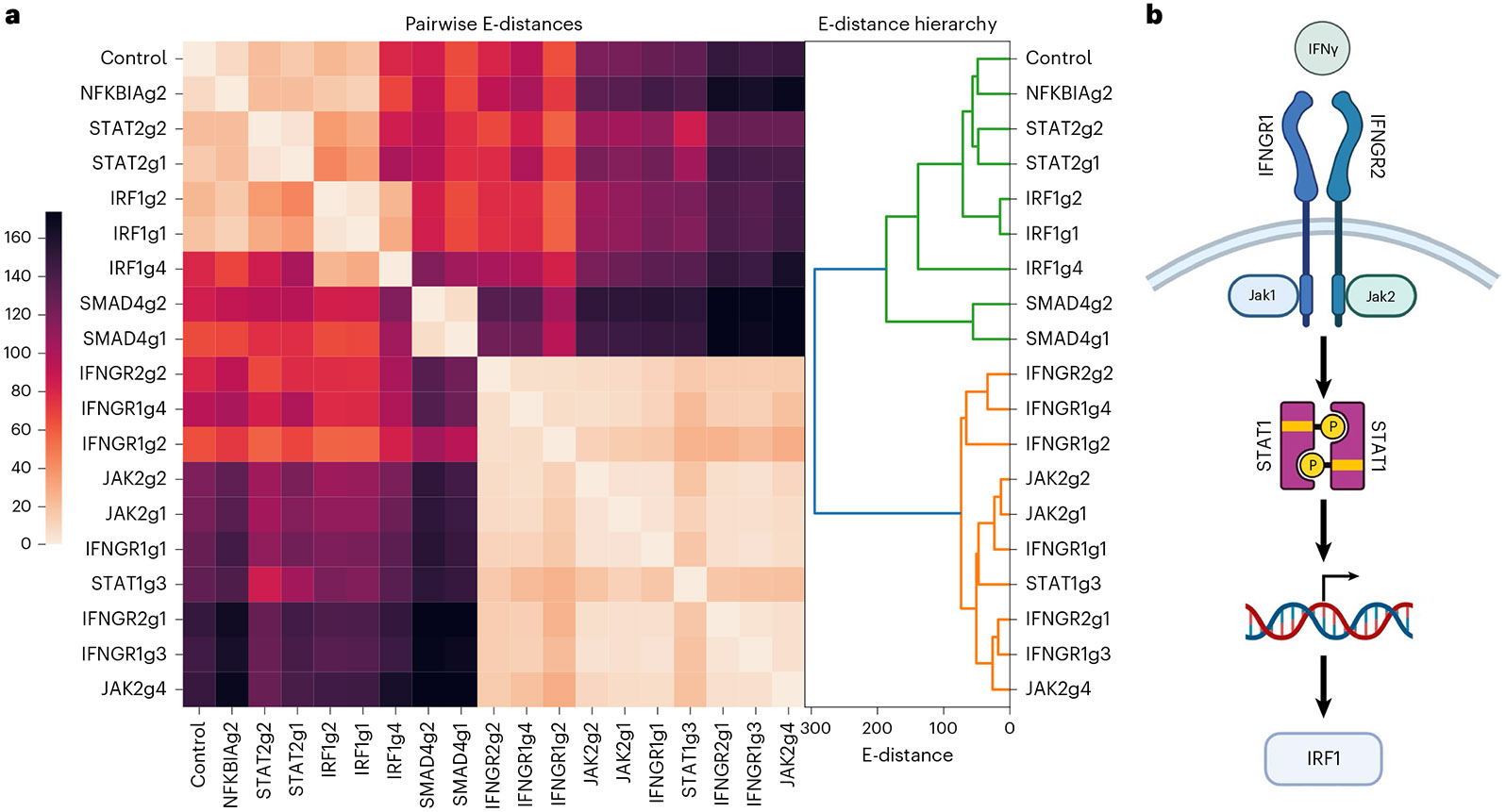
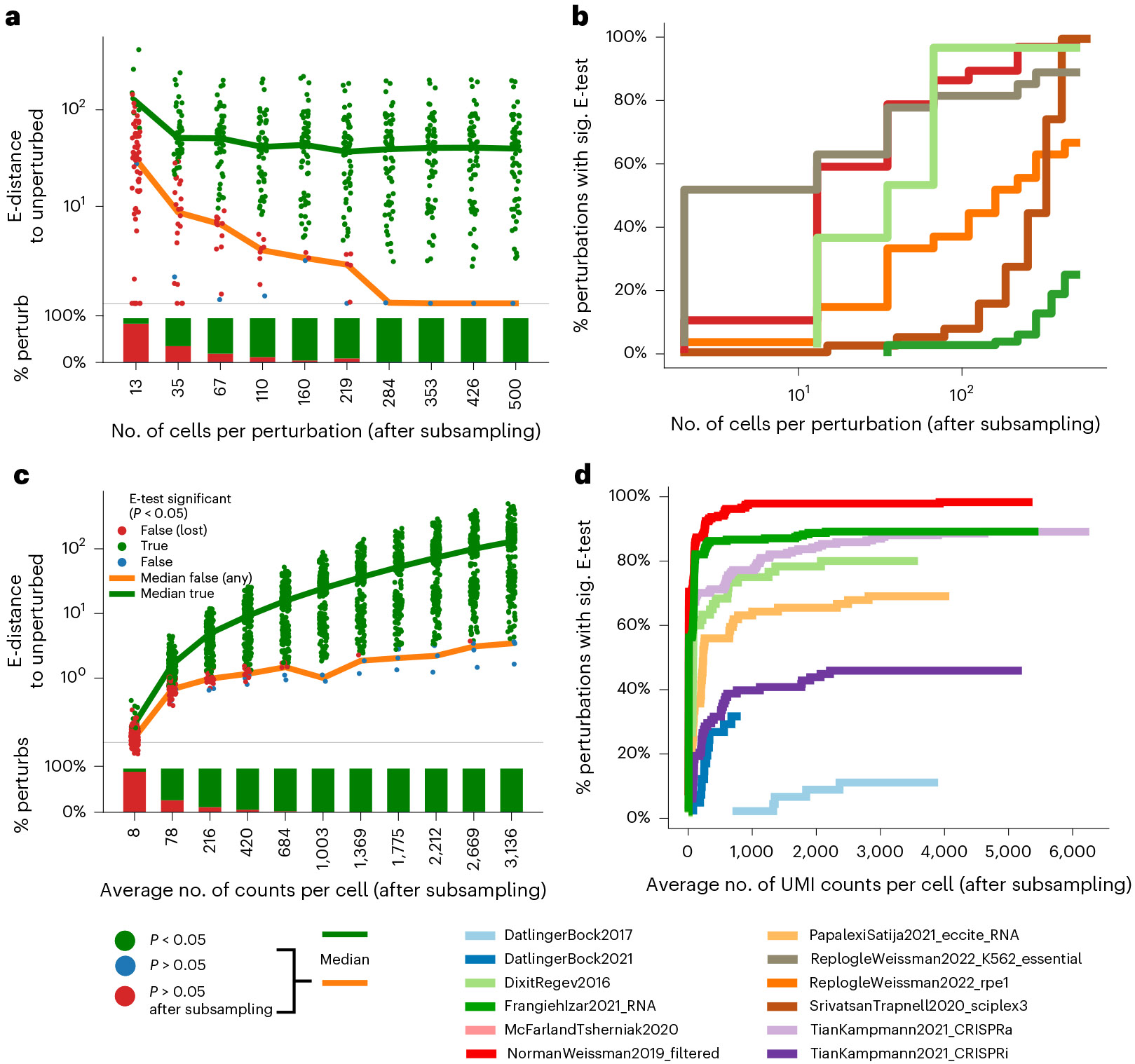
Analysis across a growing number of single-cell perturbation datasets is hampered by poor data interoperability. To facilitate development and benchmarking of computational methods, we collect a set of 44 publicly available single-cell perturbation-response datasets with molecular readouts, including transcriptomics, proteomics and epigenomics. We apply uniform quality control pipelines and harmonize feature annotations. The resulting information resource, scPerturb, enables development and testing of computational methods, and facilitates comparison and integration across datasets. We describe energy statistics (E-statistics) for quantification of perturbation effects and significance testing, and demonstrate E-distance as a general distance measure between sets of single-cell expression profiles. We illustrate the application of E-statistics for quantifying similarity and efficacy of perturbations. The perturbation-response datasets and E-statistics computation software are publicly available at scperturb.org. This work provides an information resource for researchers working with single-cell perturbation data and recommendations for experimental design, including optimal cell counts and read depth.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
-
- Jaitin DA et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896 (2016). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
