

Co-linear chaining on pangenome graphs
- PMID: 38279113
- PMCID: PMC11288099
- DOI: 10.1186/s13015-024-00250-w
Co-linear chaining on pangenome graphs
Abstract
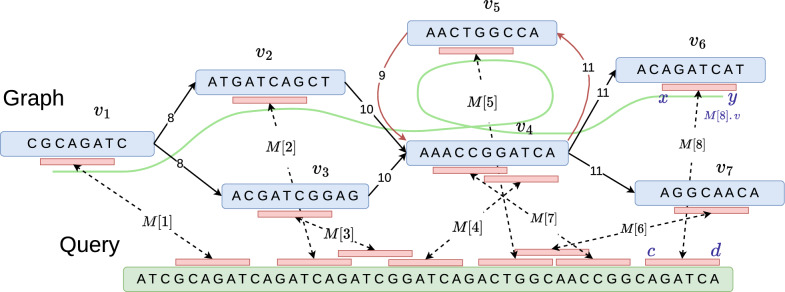
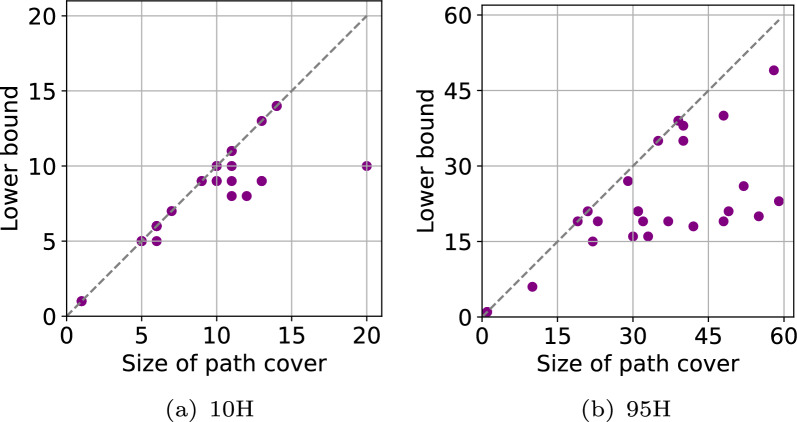
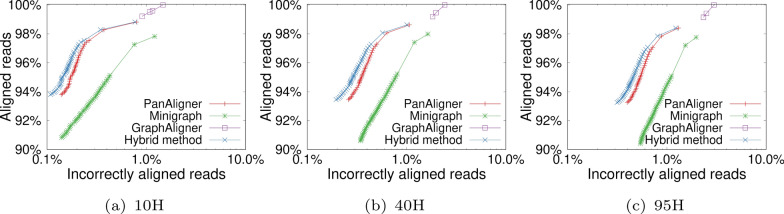
Pangenome reference graphs are useful in genomics because they compactly represent the genetic diversity within a species, a capability that linear references lack. However, efficiently aligning sequences to these graphs with complex topology and cycles can be challenging. The seed-chain-extend based alignment algorithms use co-linear chaining as a standard technique to identify a good cluster of exact seed matches that can be combined to form an alignment. Recent works show how the co-linear chaining problem can be efficiently solved for acyclic pangenome graphs by exploiting their small width and how incorporating gap cost in the scoring function improves alignment accuracy. However, it remains open on how to effectively generalize these techniques for general pangenome graphs which contain cycles. Here we present the first practical formulation and an exact algorithm for co-linear chaining on cyclic pangenome graphs. We rigorously prove the correctness and computational complexity of the proposed algorithm. We evaluate the empirical performance of our algorithm by aligning simulated long reads from the human genome to a cyclic pangenome graph constructed from 95 publicly available haplotype-resolved human genome assemblies. While the existing heuristic-based algorithms are faster, the proposed algorithm provides a significant advantage in terms of accuracy. Implementation ( https://github.com/at-cg/PanAligner ).
Keywords: Genome sequencing; Path cover; Sequence alignment; Variation graph.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
LinkOut - more resources
Full Text Sources
Miscellaneous
