

LUSTR: a new customizable tool for calling genome-wide germline and somatic short tandem repeat variants
- PMID: 38279154
- PMCID: PMC10811831
- DOI: 10.1186/s12864-023-09935-9
LUSTR: a new customizable tool for calling genome-wide germline and somatic short tandem repeat variants
Abstract
Background: Short tandem repeats (STRs) are widely distributed across the human genome and are associated with numerous neurological disorders. However, the extent that STRs contribute to disease is likely under-estimated because of the challenges calling these variants in short read next generation sequencing data. Several computational tools have been developed for STR variant calling, but none fully address all of the complexities associated with this variant class.
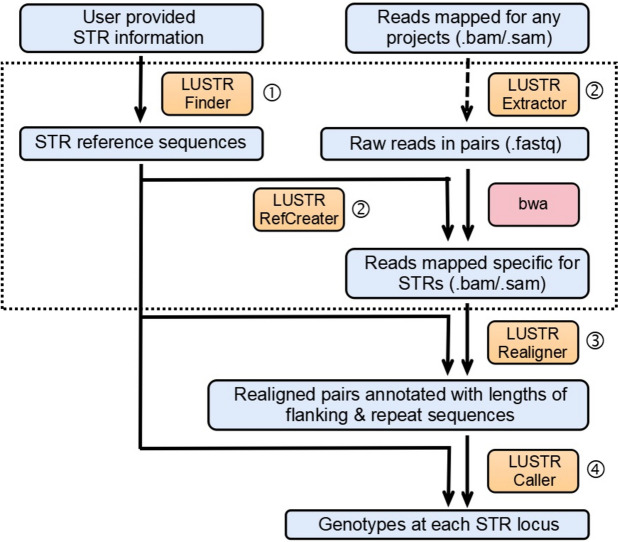
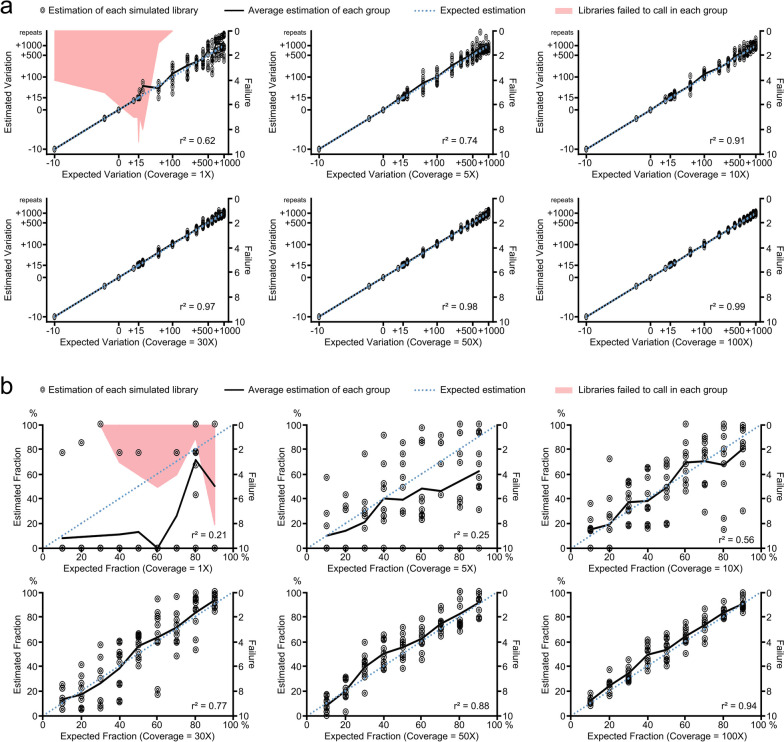
Results: Here we introduce LUSTR which is designed to address some of the challenges associated with STR variant calling by enabling more flexibility in defining STR loci, allowing for customizable modules to tailor analyses, and expanding the capability to call somatic and multiallelic STR variants. LUSTR is a user-friendly and easily customizable tool for targeted or unbiased genome-wide STR variant screening that can use either predefined or novel genome builds. Using both simulated and real data sets, we demonstrated that LUSTR accurately infers germline and somatic STR expansions in individuals with and without diseases.
Conclusions: LUSTR offers a powerful and user-friendly approach that allows for the identification of STR variants and can facilitate more comprehensive studies evaluating the role of pathogenic STR variants across human diseases.
Keywords: Bioinformatics; LUSTR; Short tandem repeats; Somatic; Variant calling tool kit.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
