

Lineage and ecology define liver tumor evolution in response to treatment
- PMID: 38280378
- PMCID: PMC10897542
- DOI: 10.1016/j.xcrm.2024.101394
Lineage and ecology define liver tumor evolution in response to treatment
Abstract
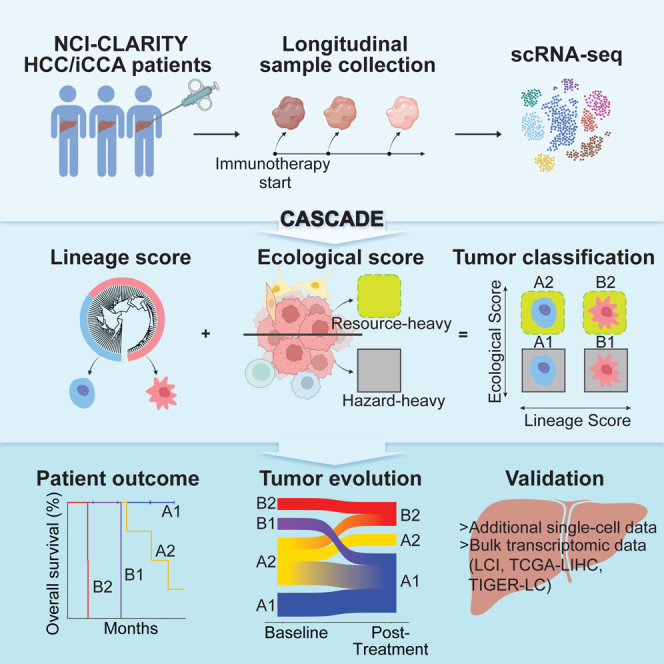
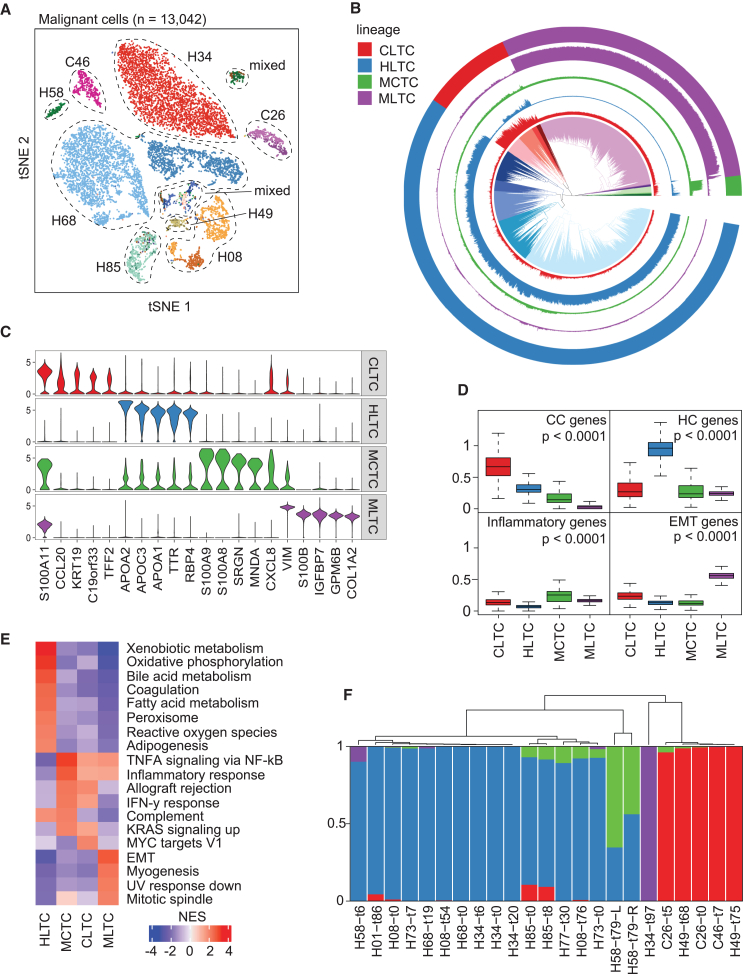
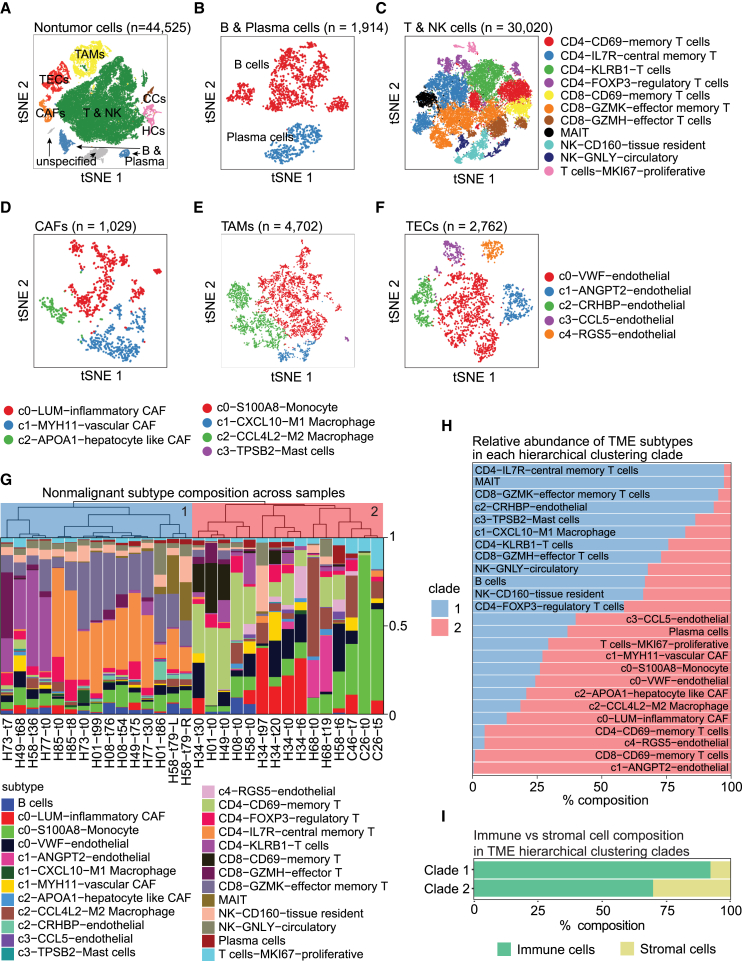
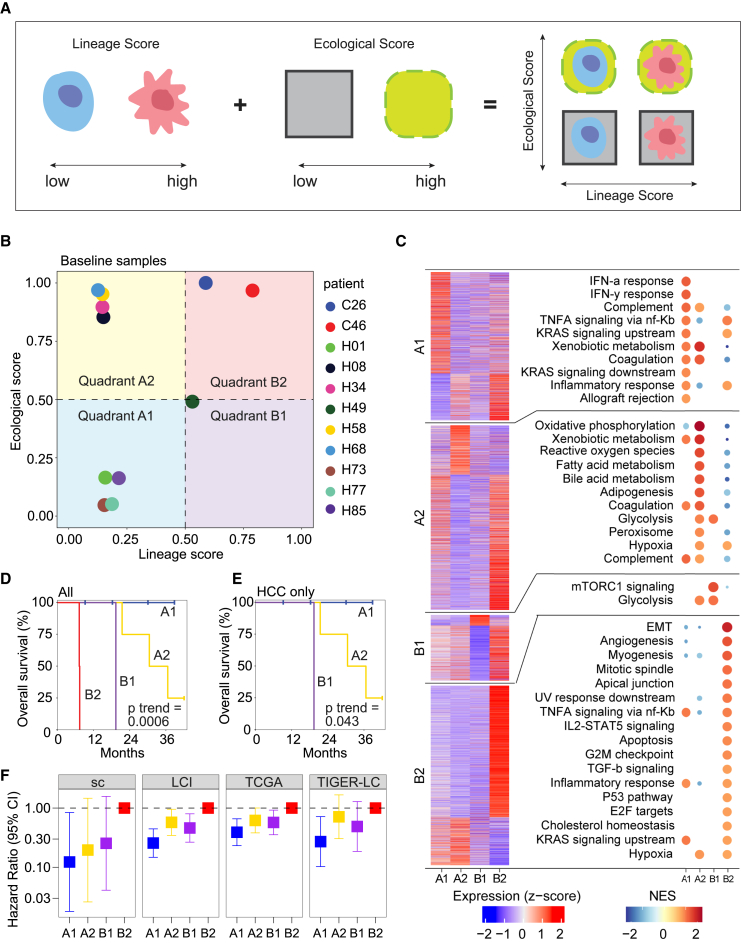
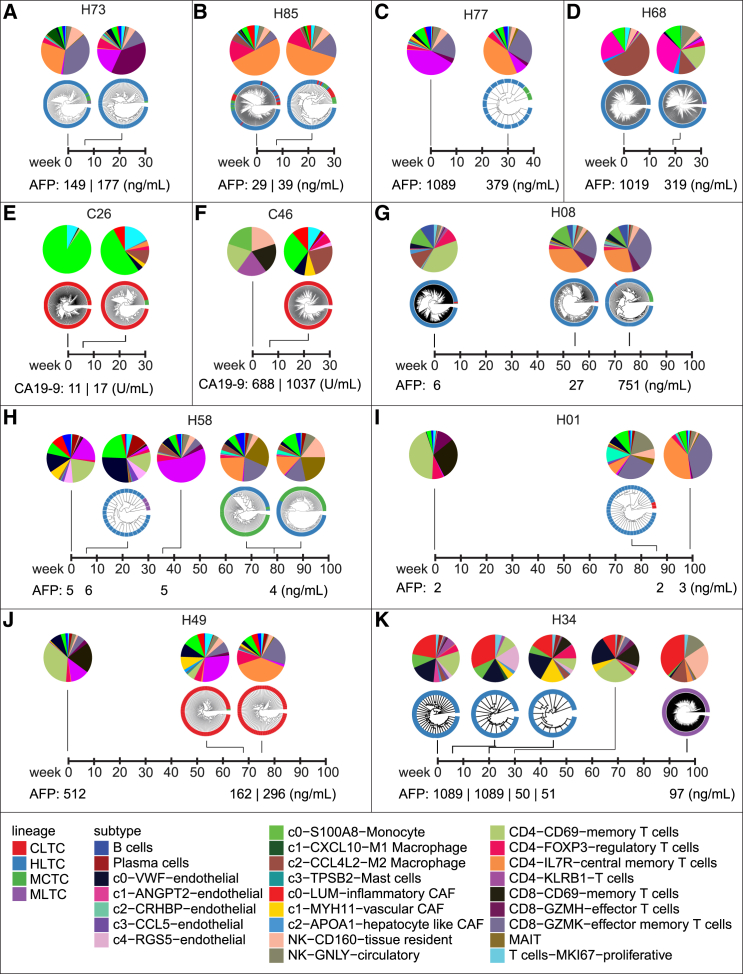
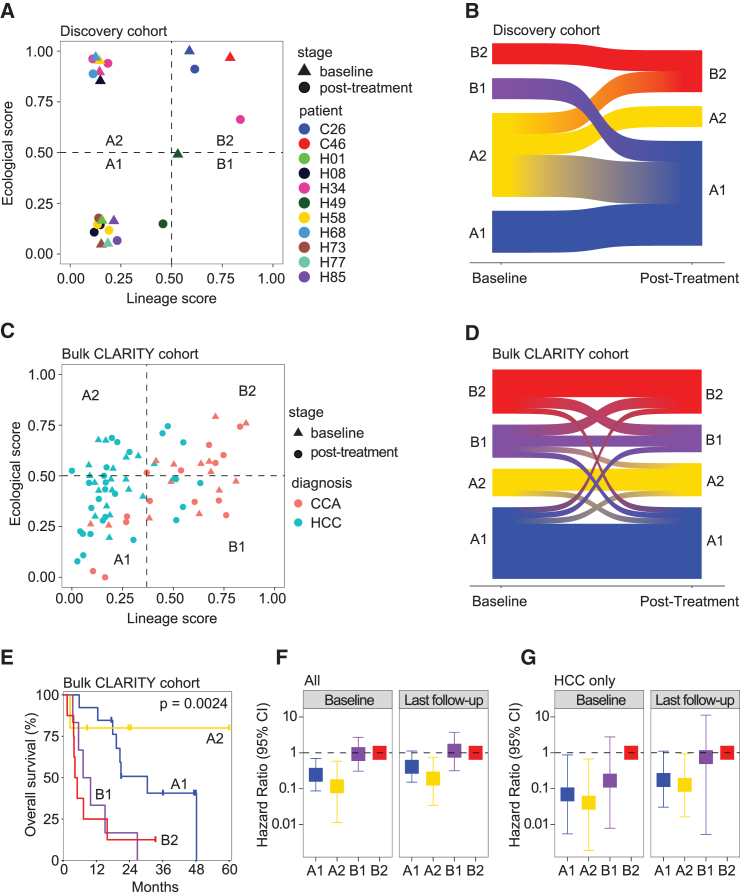
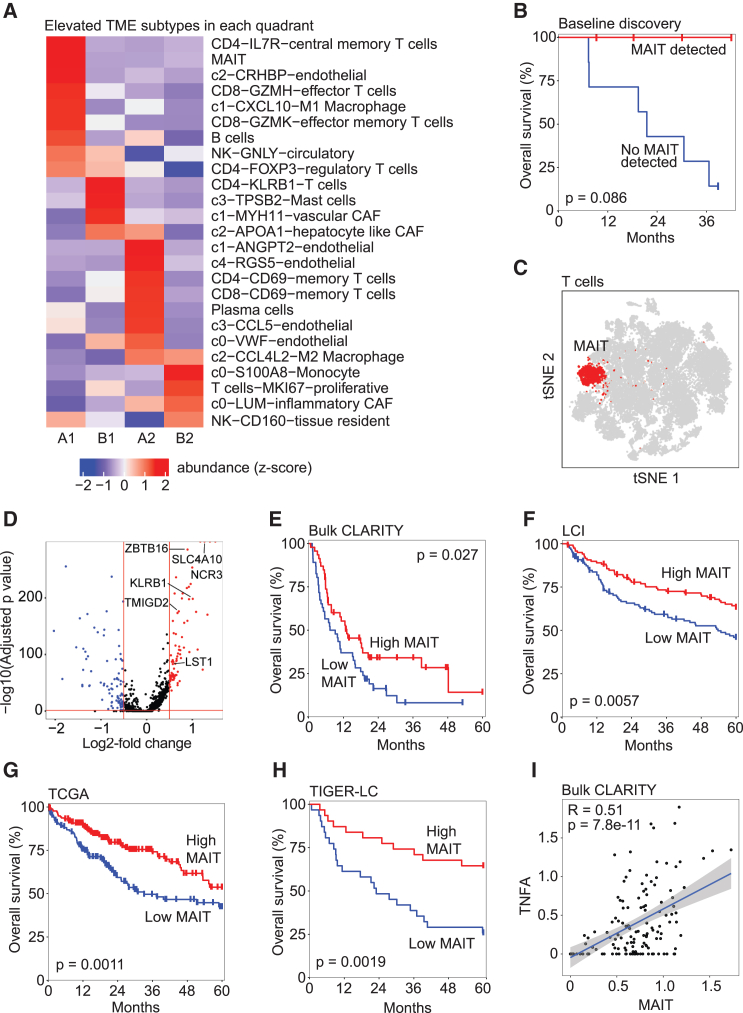
A tumor ecosystem constantly evolves over time in the face of immune predation or therapeutic intervention, resulting in treatment failure and tumor progression. Here, we present a single-cell transcriptome-based strategy to determine the evolution of longitudinal tumor biopsies from liver cancer patients by measuring cellular lineage and ecology. We construct a lineage and ecological score as joint dynamics of tumor cells and their microenvironments. Tumors may be classified into four main states in the lineage-ecological space, which are associated with clinical outcomes. Analysis of longitudinal samples reveals the evolutionary trajectory of tumors in response to treatment. We validate the lineage-ecology-based scoring system in predicting clinical outcomes using bulk transcriptomic data of additional cohorts of 716 liver cancer patients. Our study provides a framework for monitoring tumor evolution in response to therapeutic intervention.
Keywords: cholangiocarcinoma; hepatocellular carcinoma; immunotherapy; liver cancer; single cell; tumor ecology; tumor evolution; tumor heterogeneity; tumor lineage; tumor microenvironment.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Polyak K., Haviv I., Campbell I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. - PubMed
-
- Merlo L.M.F., Pepper J.W., Reid B.J., Maley C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer. 2006;6:924–935. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
