

Targeting cancer stem cell OXPHOS with tailored ruthenium complexes as a new anti-cancer strategy
- PMID: 38281027
- PMCID: PMC10821268
- DOI: 10.1186/s13046-023-02931-7
Targeting cancer stem cell OXPHOS with tailored ruthenium complexes as a new anti-cancer strategy
Abstract
Background: Previous studies by our group have shown that oxidative phosphorylation (OXPHOS) is the main pathway by which pancreatic cancer stem cells (CSCs) meet their energetic requirements; therefore, OXPHOS represents an Achille's heel of these highly tumorigenic cells. Unfortunately, therapies that target OXPHOS in CSCs are lacking.
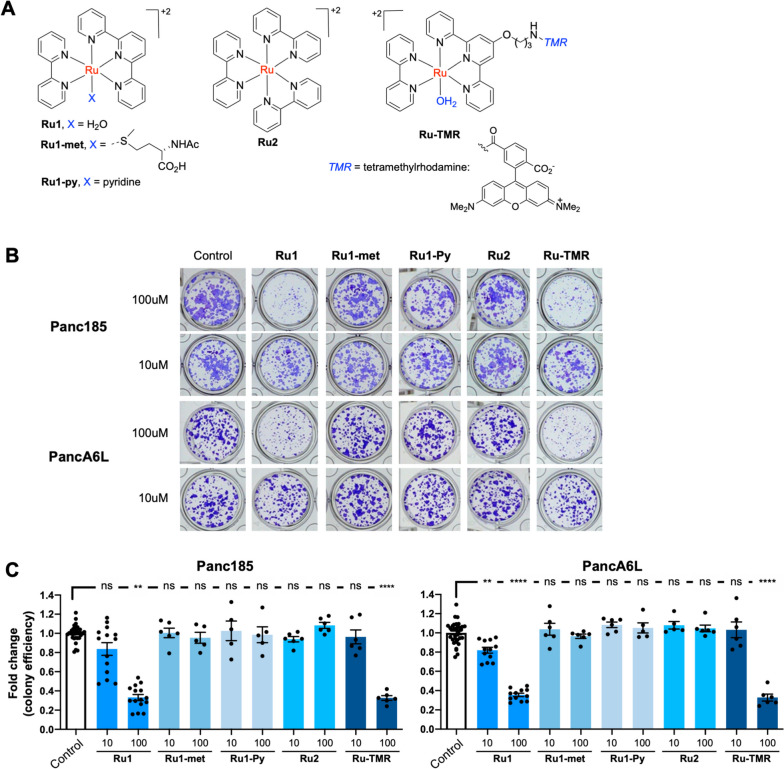
Methods: The safety and anti-CSC activity of a ruthenium complex featuring bipyridine and terpyridine ligands and one coordination labile position (Ru1) were evaluated across primary pancreatic cancer cultures and in vivo, using 8 patient-derived xenografts (PDXs). RNAseq analysis followed by mitochondria-specific molecular assays were used to determine the mechanism of action.
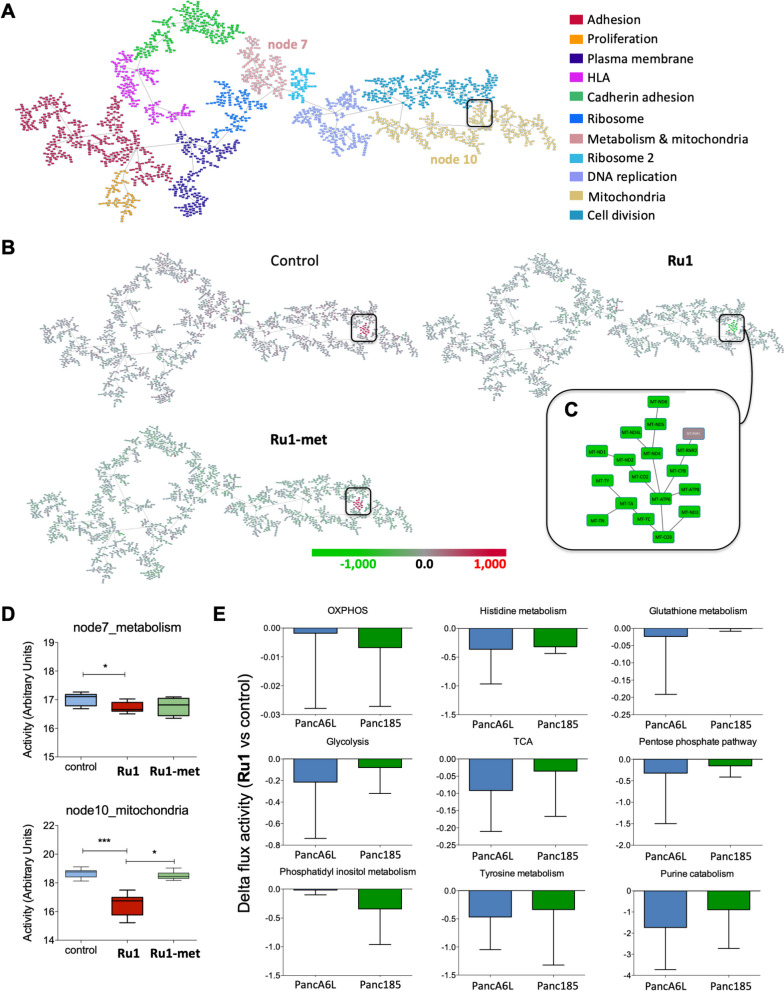
Results: We show that Ru1 is capable of inhibiting CSC OXPHOS function in vitro, and more importantly, it presents excellent anti-cancer activity, with low toxicity, across a large panel of human pancreatic PDXs, as well as in colorectal cancer and osteosarcoma PDXs. Mechanistic studies suggest that this activity stems from Ru1 binding to the D-loop region of the mitochondrial DNA of CSCs, inhibiting OXPHOS complex-associated transcription, leading to reduced mitochondrial oxygen consumption, membrane potential, and ATP production, all of which are necessary for CSCs, which heavily depend on mitochondrial respiration.
Conclusions: Overall, the coordination complex Ru1 represents not only an exciting new anti-cancer agent, but also a molecular tool to dissect the role of OXPHOS in CSCs. Results indicating that the compound is safe, non-toxic and highly effective in vivo are extremely exciting, and have allowed us to uncover unprecedented mechanistic possibilities to fight different cancer types based on targeting CSC OXPHOS.
Keywords: Anti-cancer agents; Cancer stem cells; Colon cancer; Mitochondrial DNA; Oxidative phosphorylation; Pancreatic ductal adenocarcinoma; Patient-derived xenografts; Ruthenium complexes.
© 2024. The Author(s).
Conflict of interest statement
A.G-P and J.A.F.V. are shareholders of Biomedica Molecular Medicine SL. The
Figures








Similar articles
-
Targeting flavin-containing enzymes eliminates cancer stem cells (CSCs), by inhibiting mitochondrial respiration: Vitamin B2 (Riboflavin) in cancer therapy.Aging (Albany NY). 2017 Dec 16;9(12):2610-2628. doi: 10.18632/aging.101351. Aging (Albany NY). 2017. PMID: 29253841 Free PMC article.
-
Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells.Nat Commun. 2020 Oct 16;11(1):5265. doi: 10.1038/s41467-020-18954-z. Nat Commun. 2020. PMID: 33067432 Free PMC article.
-
Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells.Oncotarget. 2016 Jun 7;7(23):34084-99. doi: 10.18632/oncotarget.9122. Oncotarget. 2016. PMID: 27136895 Free PMC article.
-
OXPHOS-targeting drugs in oncology: new perspectives.Expert Opin Ther Targets. 2023 Jul-Dec;27(10):939-952. doi: 10.1080/14728222.2023.2261631. Epub 2023 Oct 30. Expert Opin Ther Targets. 2023. PMID: 37736880 Free PMC article. Review.
-
Pancreatic cancer stem cells: A state or an entity?Semin Cancer Biol. 2018 Dec;53:223-231. doi: 10.1016/j.semcancer.2018.08.007. Epub 2018 Aug 18. Semin Cancer Biol. 2018. PMID: 30130664 Review.
Cited by
-
Everything you ever wanted to know about cancer stem cells in neuroendocrine neoplasms but were afraid to ask.Endocr Oncol. 2024 Dec 19;4(1):e240006. doi: 10.1530/EO-24-0006. eCollection 2024 Jan 1. Endocr Oncol. 2024. PMID: 39822777 Free PMC article. Review.
-
Preclinical Trials of Cancer Stem Cells Targeted by Metal-Based Coordination Complexes: A Systematic Review.Pharmaceutics. 2025 Jul 18;17(7):931. doi: 10.3390/pharmaceutics17070931. Pharmaceutics. 2025. PMID: 40733139 Free PMC article. Review.
-
Metabolic plasticity in pancreatic cancer: The mitochondrial connection.Mol Metab. 2025 Feb;92:102089. doi: 10.1016/j.molmet.2024.102089. Epub 2024 Dec 28. Mol Metab. 2025. PMID: 39736443 Free PMC article. Review.
-
Platinum iodido drugs show potential anti-tumor activity, affecting cancer cell metabolism and inducing ROS and senescence in gastrointestinal cancer cells.Commun Biol. 2024 Mar 22;7(1):353. doi: 10.1038/s42003-024-06052-5. Commun Biol. 2024. PMID: 38519773 Free PMC article.
-
Reprogramming of glucose metabolism in pancreatic cancer: mechanisms, implications, and therapeutic perspectives.Front Immunol. 2025 Jun 24;16:1586959. doi: 10.3389/fimmu.2025.1586959. eCollection 2025. Front Immunol. 2025. PMID: 40630951 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
- RYC-2012-12104/Ministerio de Ciencia e Innovación
- PID2019-110320RB-I00/Ministerio de Ciencia e Innovación
- PID2019-108624RB-I00/Ministerio de Ciencia e Innovación
- PDC2021-121508-I00/Ministerio de Ciencia e Innovación
- CTQ2016-81797-REDC/Ministerio de Ciencia e Innovación
- GC16173694BARB/Fundación Científica Asociación Española Contra el Cáncer
- IDEAS222917FERN/Fundación Científica Asociación Española Contra el Cáncer
- PI18/00757/Instituto de Salud Carlos III
- PI21/01110/Instituto de Salud Carlos III
- PT20/00045/Instituto de Salud Carlos III
- 340055/H2020 European Research Council
- 899334/H2020 European Research Council
- 111746/Deutsche Kinderkrebsstiftung
- CRC 1279/Deutsche Bundesstiftung Umwelt
- IN855A-2018/16/Agencia Gallega de Innovación
- CC21-20122/'la Caixa' Foundation
LinkOut - more resources
Full Text Sources
Medical
