

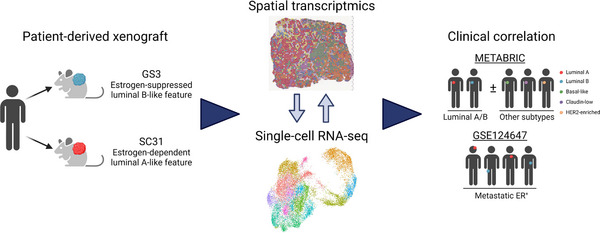
Molecular features of luminal breast cancer defined through spatial and single-cell transcriptomics
- PMID: 38282415
- PMCID: PMC10823285
- DOI: 10.1002/ctm2.1548
Molecular features of luminal breast cancer defined through spatial and single-cell transcriptomics
Abstract
Background: Intratumour heterogeneity is a hallmark of most solid tumours, including breast cancers. We applied spatial transcriptomics and single-cell RNA-sequencing on patient-derived xenografts (PDXs) to profile spatially resolved cell populations within oestrogen receptor-positive (ER+ ) breast cancer and to elucidate their importance in oestrogen-dependent tumour growth.
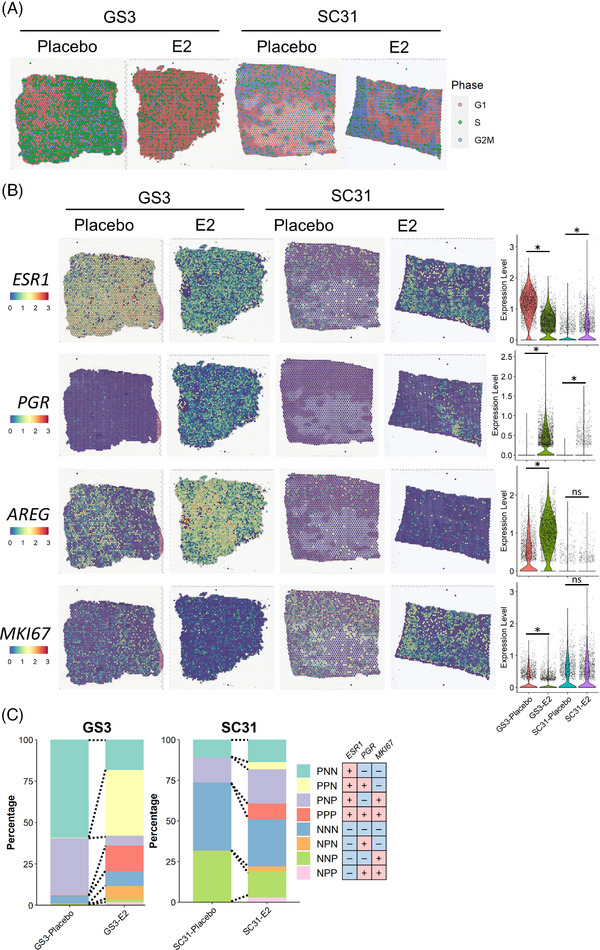
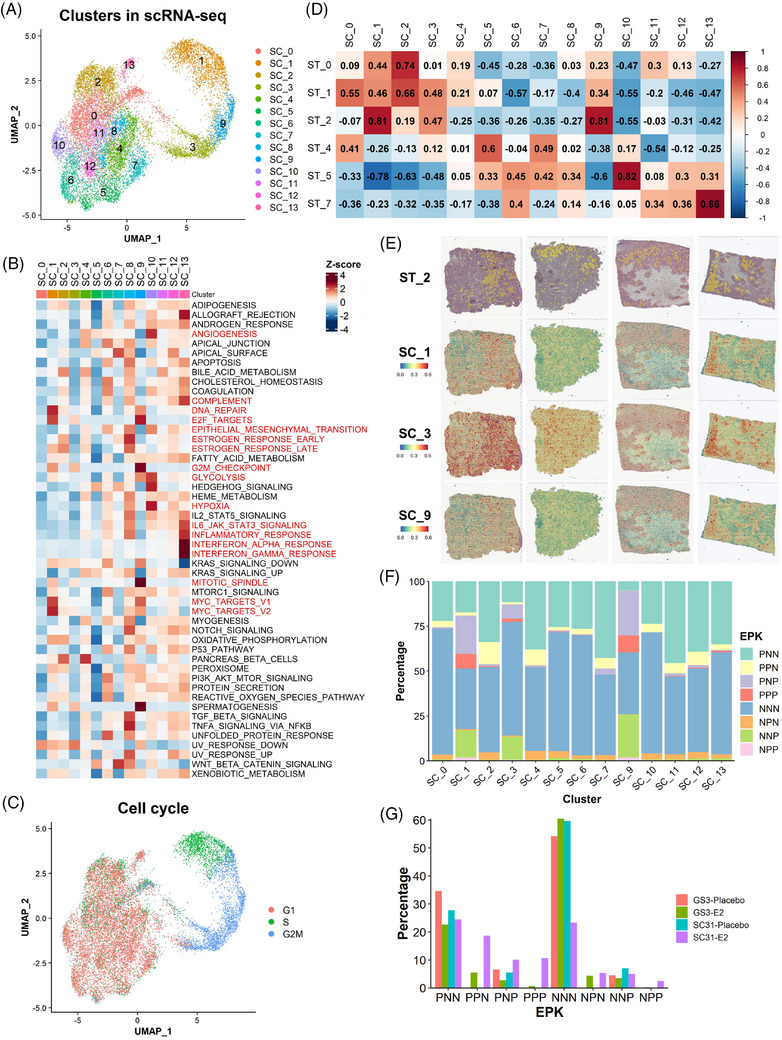
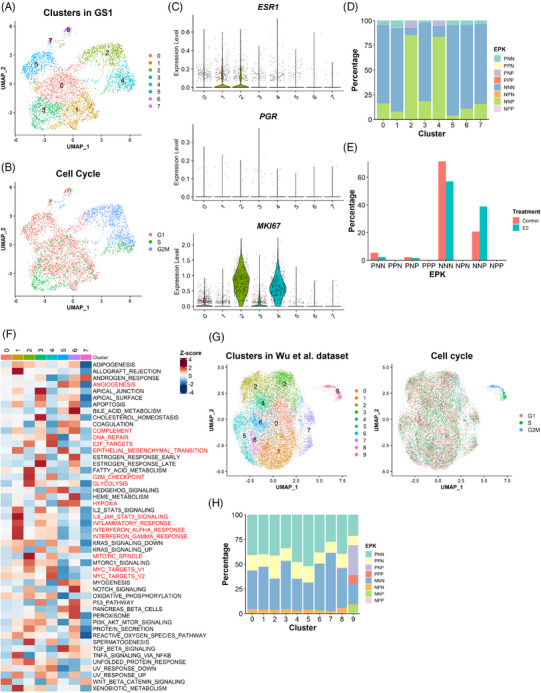
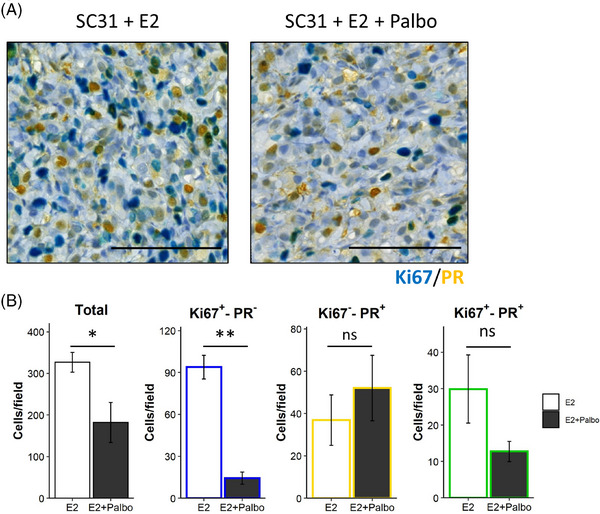
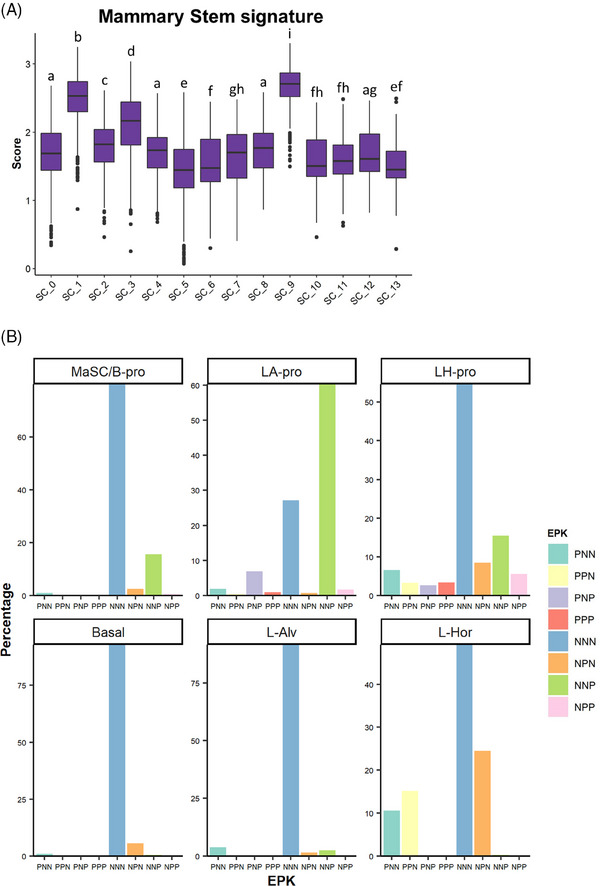
Methods: Two PDXs of 'ER-high' breast cancers with opposite oestrogen-mediated growth responses were investigated: oestrogen-suppressed GS3 (80-100% ER) and oestrogen-dependent SC31 (40-90% ER) models. The observation was validated via single-cell analyses on an 'ER-low' PDX, GS1 (5% ER). The results from our spatial and single-cell analyses were further supported by a public ER+ breast cancer single-cell dataset and protein-based dual immunohistochemistry (IHC) of SC31 examining important luminal cancer markers (i.e., ER, progesterone receptor and Ki67). The translational implication of our findings was assessed by clinical outcome analyses on publicly available cohorts.
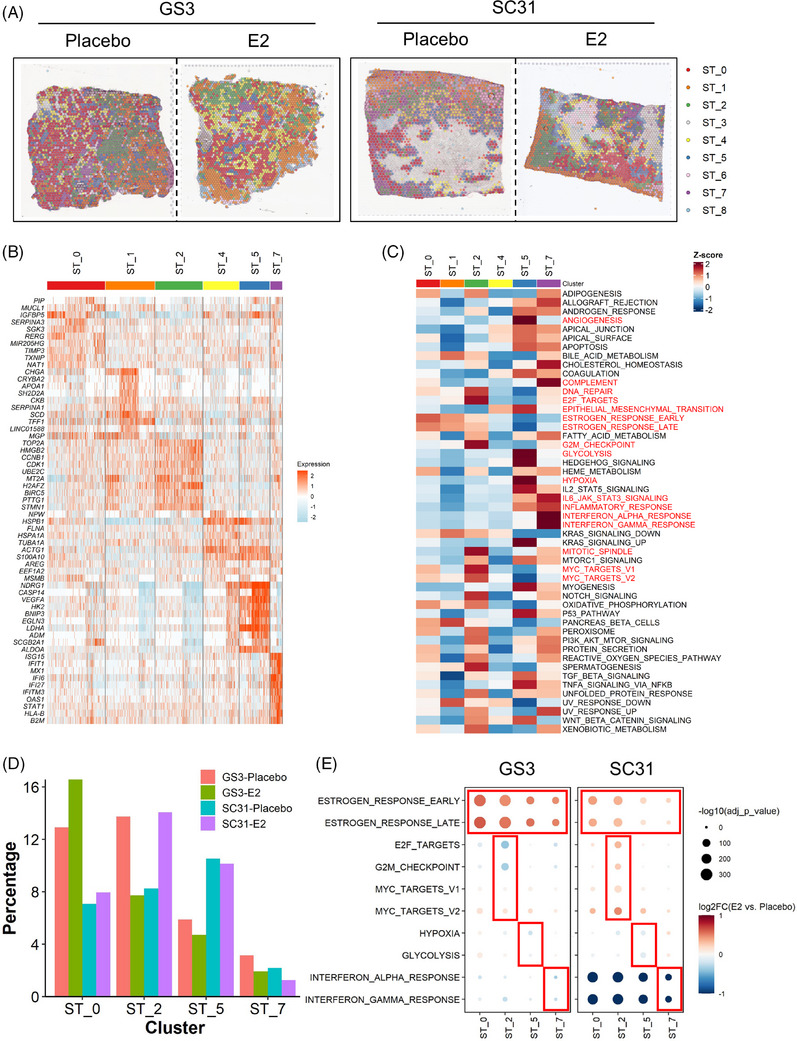
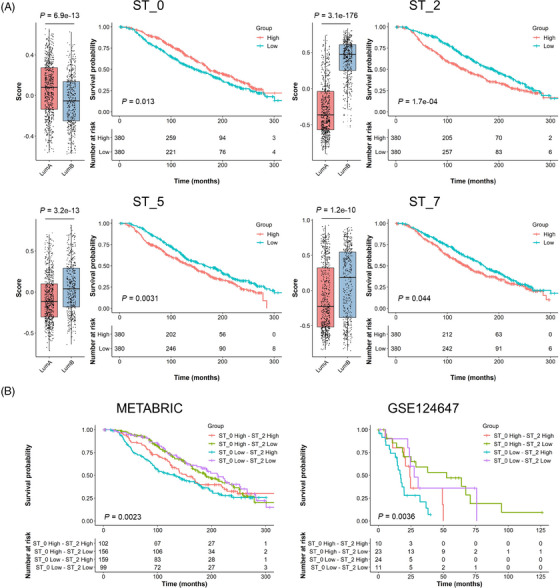
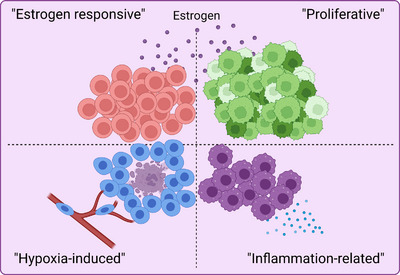
Results: Our space-gene-function study revealed four spatially distinct compartments within ER+ breast cancers. These compartments showed functional diversity (oestrogen-responsive, proliferative, hypoxia-induced and inflammation-related). The 'proliferative' population, rather than the 'oestrogen-responsive' compartment, was crucial for oestrogen-dependent tumour growth, leading to the acquisition of luminal B-like features. The cells expressing typical oestrogen-responsive genes like PGR were not directly linked to oestrogen-dependent proliferation. Dual IHC analyses demonstrated the distinct contribution of the Ki67+ proliferative cells toward oestrogen-mediated growth and their response to a CDK4/6 inhibitor. The gene signatures derived from the proliferative, hypoxia-induced and inflammation-related compartments were significantly correlated with worse clinical outcomes, while patients with the oestrogen-responsive signature showed better prognoses, suggesting that this compartment would not be directly associated with oestrogen-dependent tumour progression.
Conclusions: Our study identified the gene signature in our 'proliferative' compartment as an important determinant of luminal cancer subtypes. This 'proliferative' cell population is a causative feature of luminal B breast cancer, contributing toward its aggressive behaviours.
Keywords: breast cancer; intratumour heterogeneity; oestrogen receptor; single-cell RNA-sequencing; spatial transcriptomics.
© 2024 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures
References
-
- Perou CM, Sørile T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747‐752. - PubMed
-
- Ades F, Zardavas D, Bozovic‐Spasojevic I, et al. Luminal B breast cancer: molecular characterization, clinical management, and future perspectives. J Clin Oncol. 2014;32(25):2794‐2803. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials