

Ebola virus sequesters IRF3 in viral inclusion bodies to evade host antiviral immunity
- PMID: 38285487
- PMCID: PMC10945704
- DOI: 10.7554/eLife.88122
Ebola virus sequesters IRF3 in viral inclusion bodies to evade host antiviral immunity
Abstract
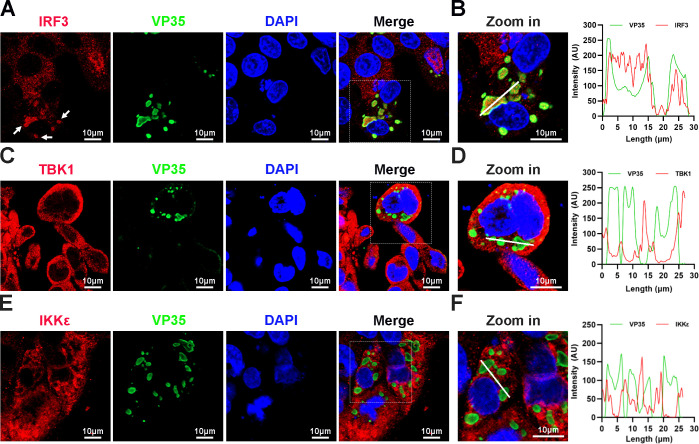
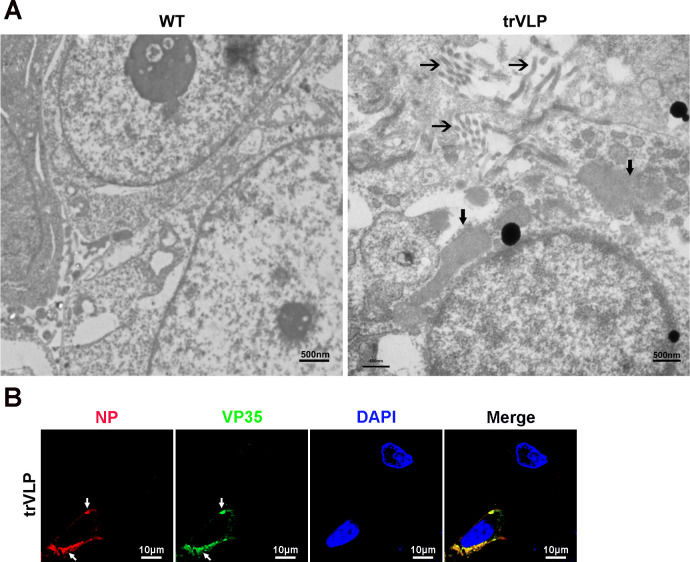
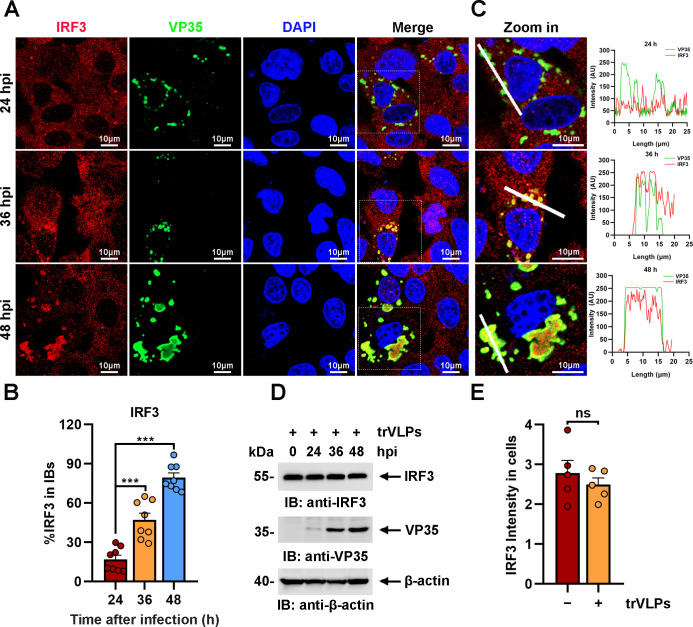
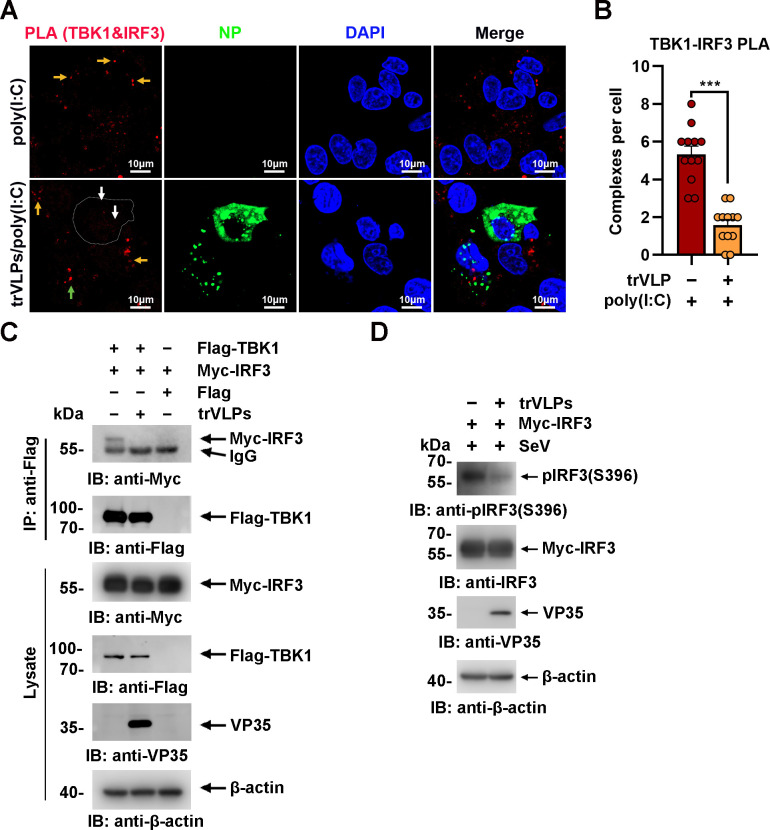
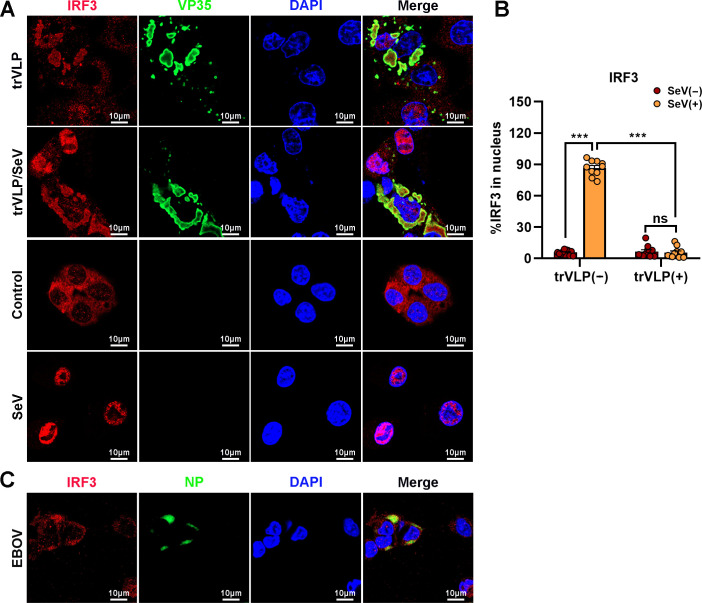
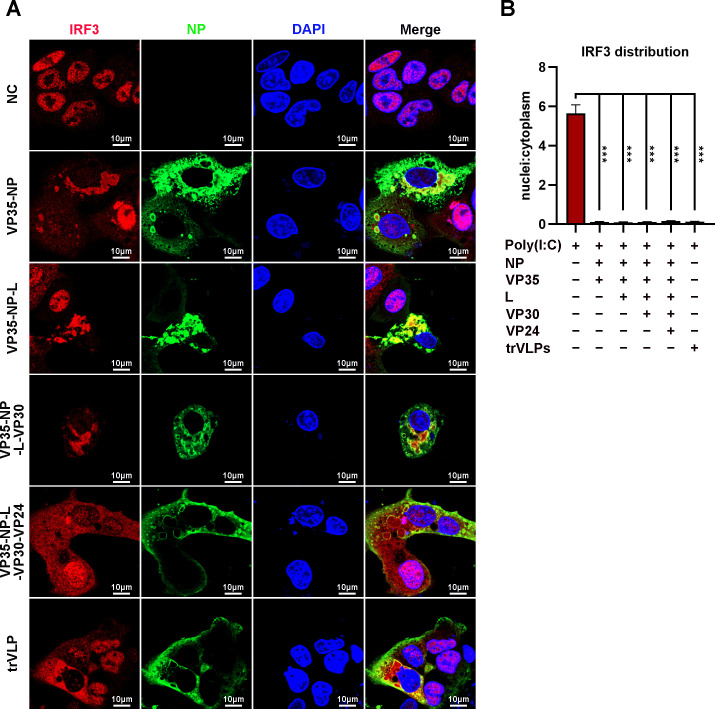
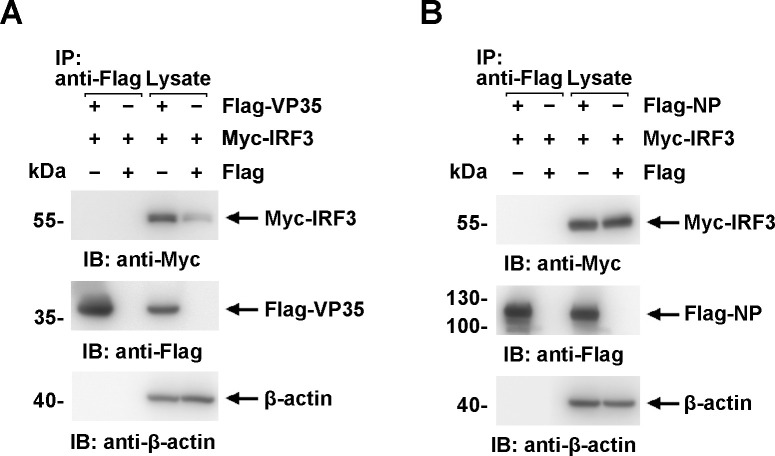
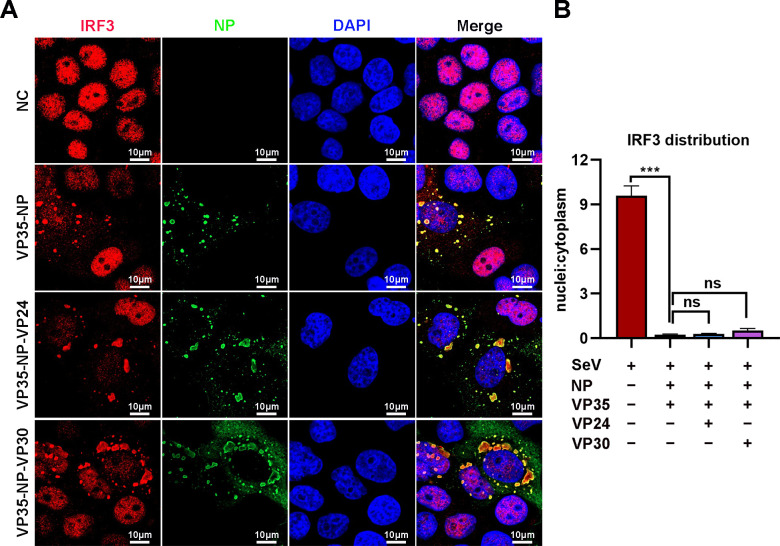
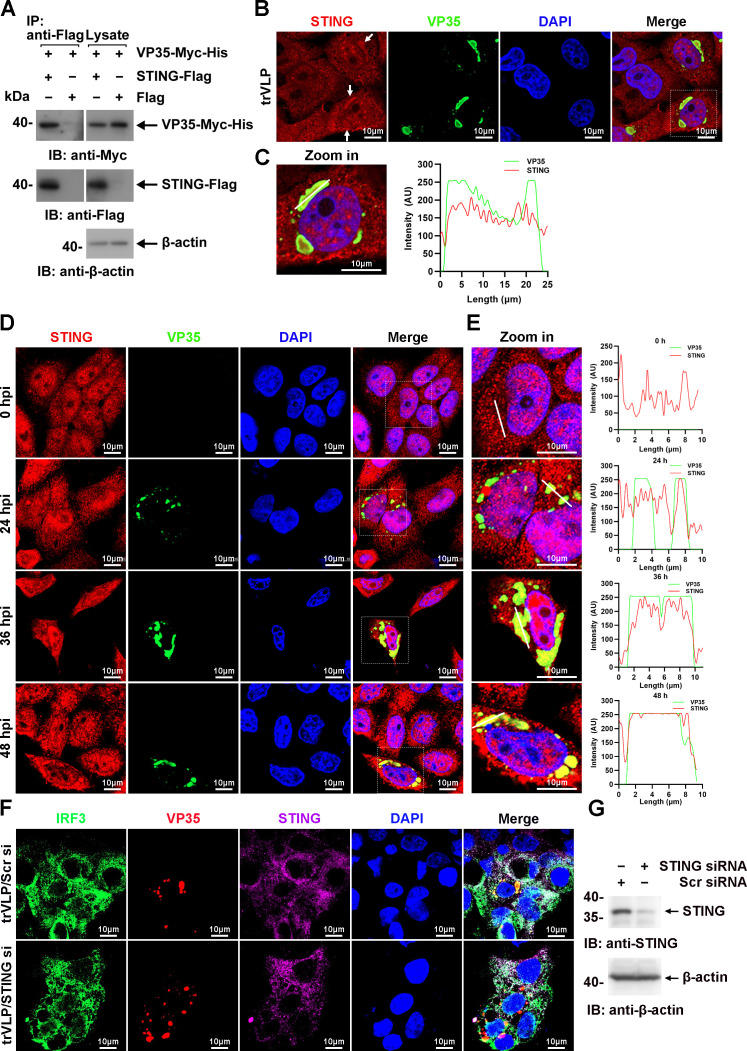
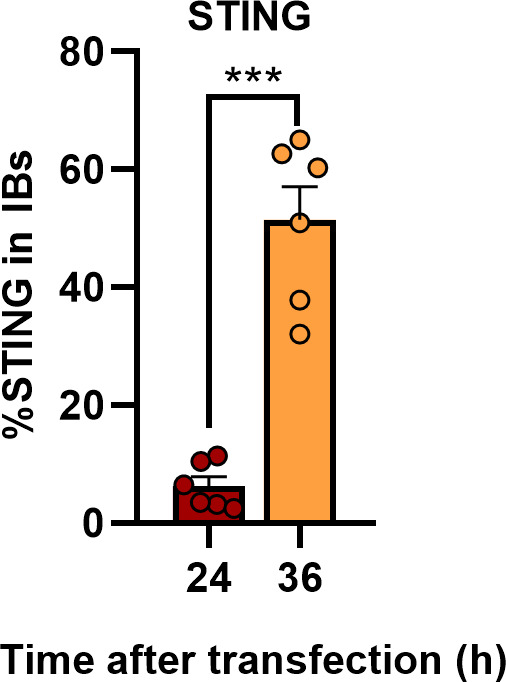
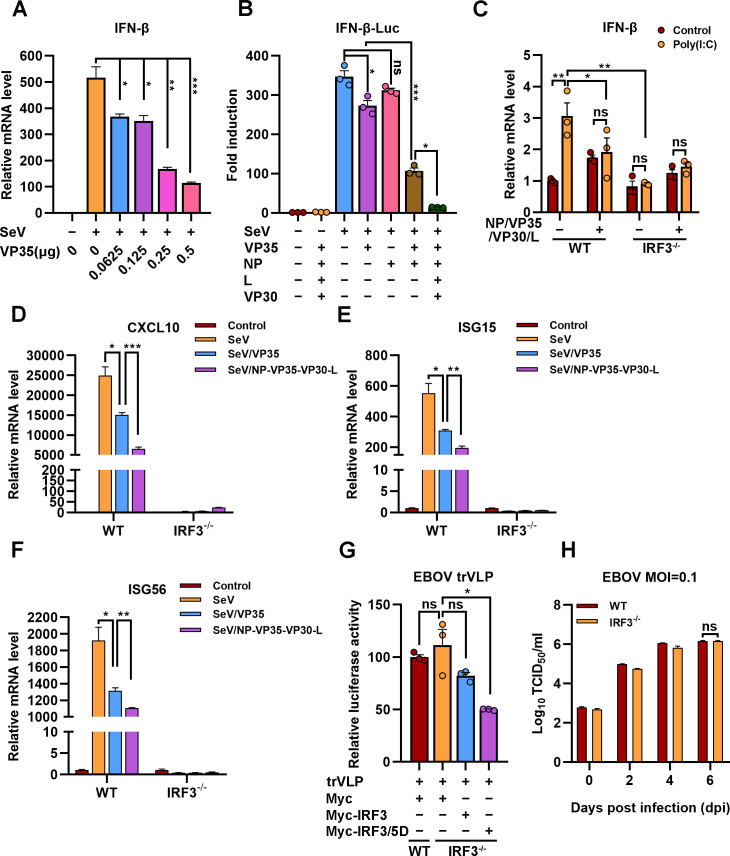
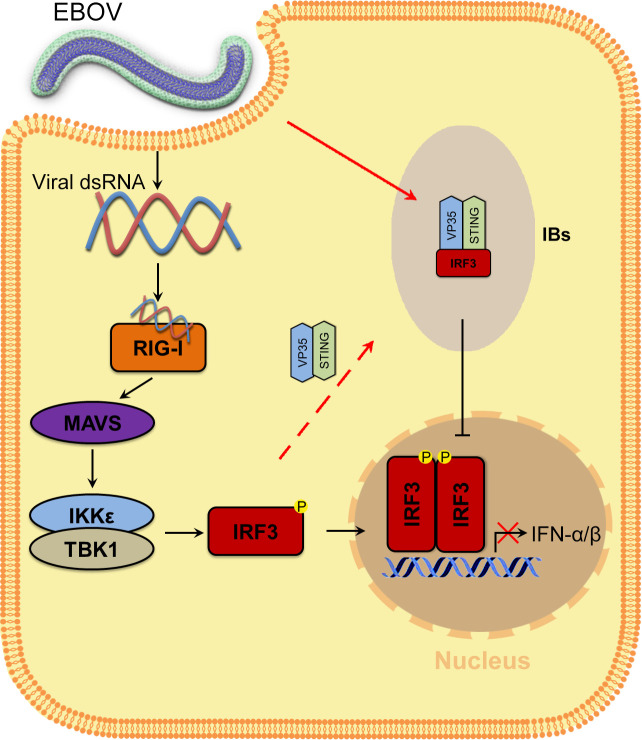
Viral inclusion bodies (IBs) commonly form during the replication of Ebola virus (EBOV) in infected cells, but their role in viral immune evasion has rarely been explored. Here, we found that interferon regulatory factor 3 (IRF3), but not TANK-binding kinase 1 (TBK1) or IκB kinase epsilon (IKKε), was recruited and sequestered in viral IBs when the cells were infected by EBOV transcription- and replication-competent virus-like particles (trVLPs). Nucleoprotein/virion protein 35 (VP35)-induced IBs formation was critical for IRF3 recruitment and sequestration, probably through interaction with STING. Consequently, the association of TBK1 and IRF3, which plays a vital role in type I interferon (IFN-I) induction, was blocked by EBOV trVLPs infection. Additionally, IRF3 phosphorylation and nuclear translocation induced by Sendai virus or poly(I:C) stimulation were suppressed by EBOV trVLPs. Furthermore, downregulation of STING significantly attenuated VP35-induced IRF3 accumulation in IBs. Coexpression of the viral proteins by which IB-like structures formed was much more potent in antagonizing IFN-I than expression of the IFN-I antagonist VP35 alone. These results suggested a novel immune evasion mechanism by which EBOV evades host innate immunity.
Keywords: Ebola virus; VP24; VP35; immunology; infectious disease; inflammation; microbiology; virus inclusion bodies; virus infection; viruses.
© 2023, Zhu et al.
Conflict of interest statement
LZ, JJ, TW, YH, HL, TG, QD, YJ, PL, ZL, YH, XL, CC No competing interests declared
Figures
Update of
- doi: 10.1101/2023.04.20.537734
- doi: 10.7554/eLife.88122.1
- doi: 10.7554/eLife.88122.2
References
MeSH terms
Substances
Grants and funding
- 82372255/National Natural Science Foundation of China
- 2022ACCP-MS04/Advanced Customer Cultivation Project of the Wuhan National Biosafety Laboratory, the Chinese Academy of Sciences
- 2018ZX09711003-005-005/National Major Science and Technology Projects of China
- 2022YFC2600704/National Major Science and Technology Projects of China
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
