

Necroptosis does not drive disease pathogenesis in a mouse infective model of SARS-CoV-2 in vivo
- PMID: 38286985
- PMCID: PMC10825138
- DOI: 10.1038/s41419-024-06471-6
Necroptosis does not drive disease pathogenesis in a mouse infective model of SARS-CoV-2 in vivo
Abstract
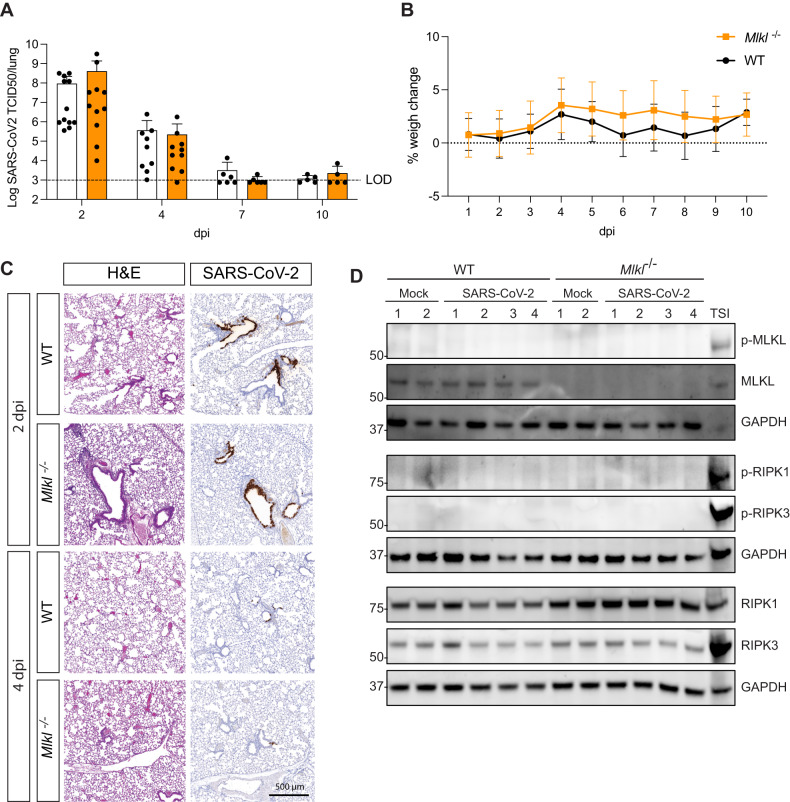
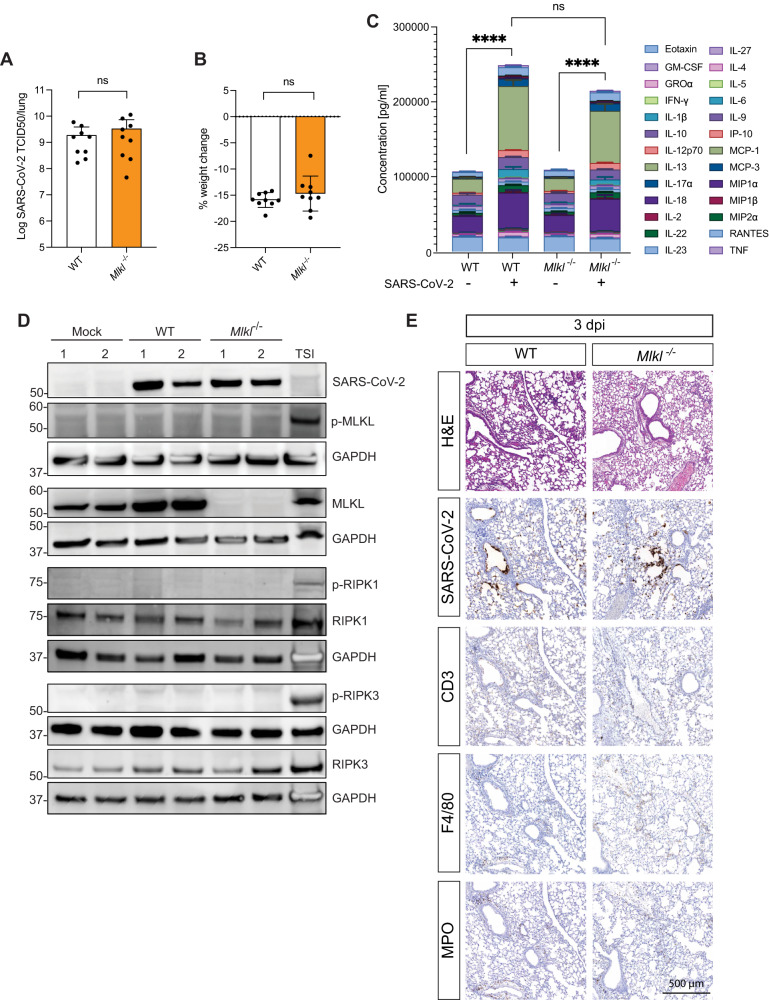
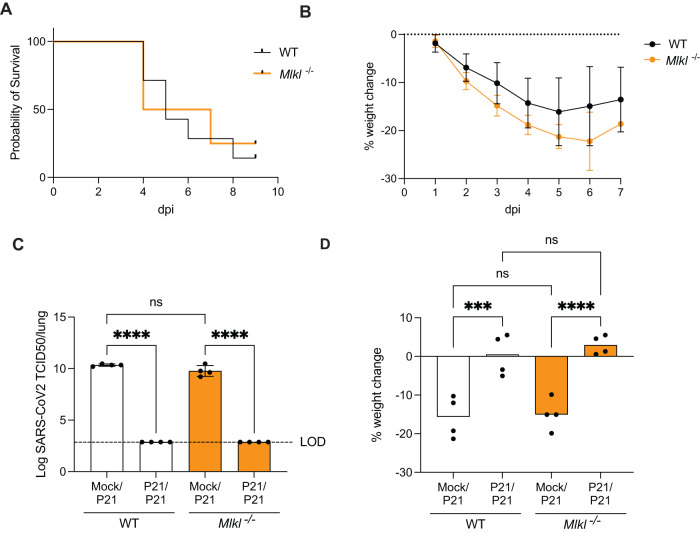
Necroptosis, a type of lytic cell death executed by the pseudokinase Mixed Lineage Kinase Domain-Like (MLKL) has been implicated in the detrimental inflammation caused by SARS-CoV-2 infection. We minimally and extensively passaged a single clinical SARS-CoV-2 isolate to create models of mild and severe disease in mice allowing us to dissect the role of necroptosis in SARS-CoV-2 disease pathogenesis. We infected wild-type and MLKL-deficient mice and found no significant differences in viral loads or lung pathology. In our model of severe COVID-19, MLKL-deficiency did not alter the host response, ameliorate weight loss, diminish systemic pro-inflammatory cytokines levels, or prevent lethality in aged animals. Our in vivo models indicate that necroptosis is dispensable in the pathogenesis of mild and severe COVID-19.
© 2024. The Author(s).
Conflict of interest statement
JMM has received research funding from Anaxis Pharma Pty Ltd. All other authors declare no competing interests.
Figures
References
-
- Singh S, McNab C, Olson RM, Bristol N, Nolan C, Bergstrøm E, et al. How an outbreak became a pandemic: a chronological analysis of crucial junctures and international obligations in the early months of the COVID-19 pandemic. Lancet. 2021;398:2109–24. doi: 10.1016/S0140-6736(21)01897-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
