

Evaluating protein cross-linking as a therapeutic strategy to stabilize SOD1 variants in a mouse model of familial ALS
- PMID: 38289969
- PMCID: PMC10826971
- DOI: 10.1371/journal.pbio.3002462
Evaluating protein cross-linking as a therapeutic strategy to stabilize SOD1 variants in a mouse model of familial ALS
Abstract
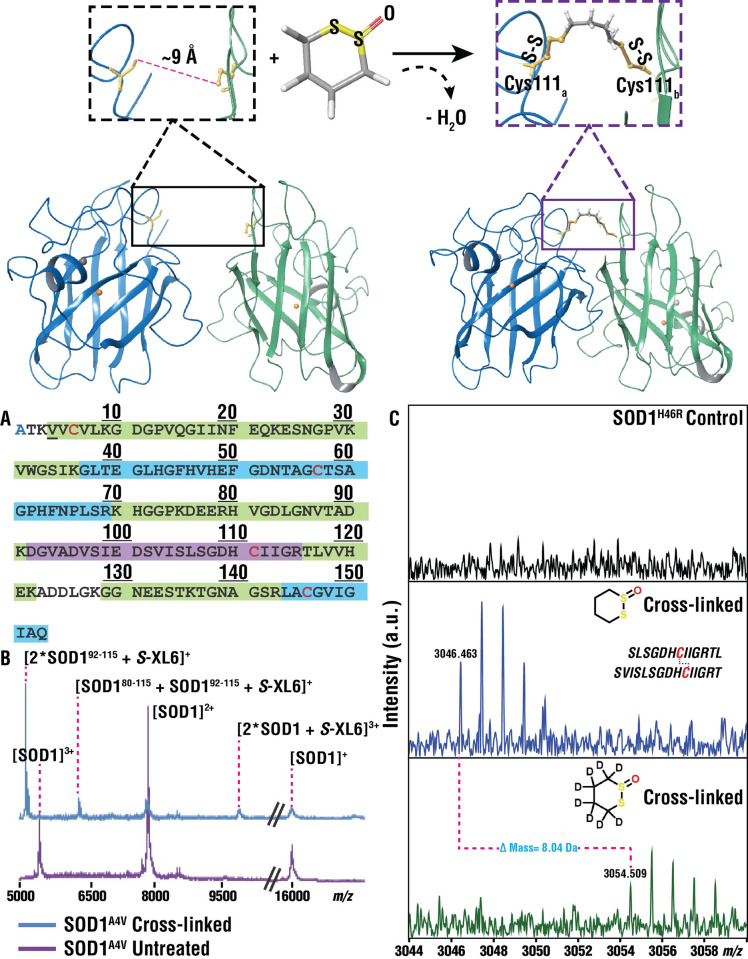
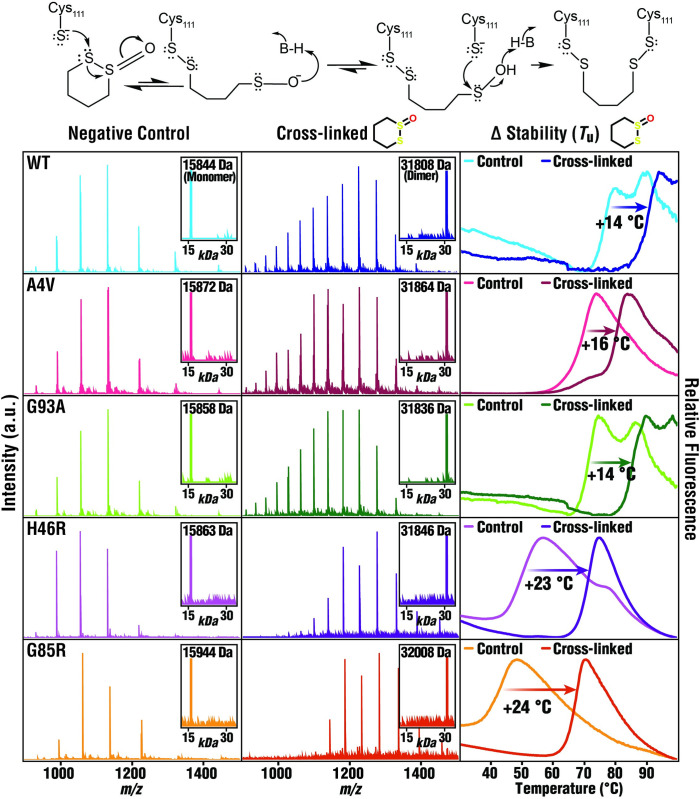
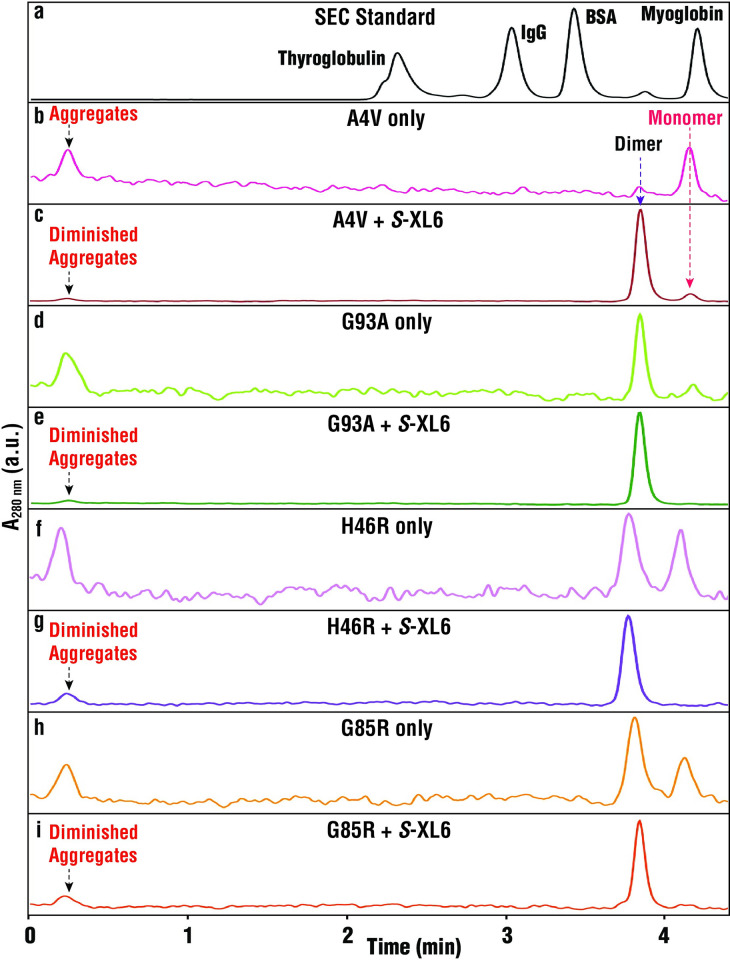
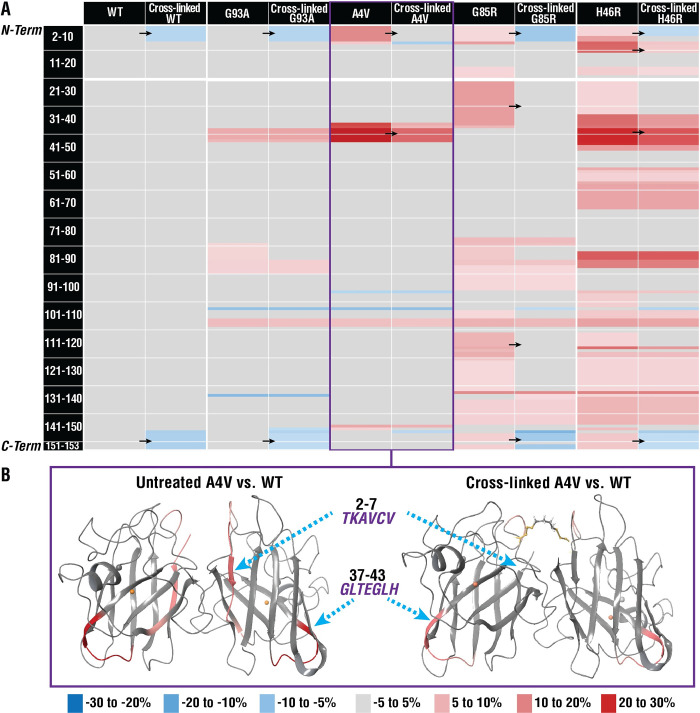
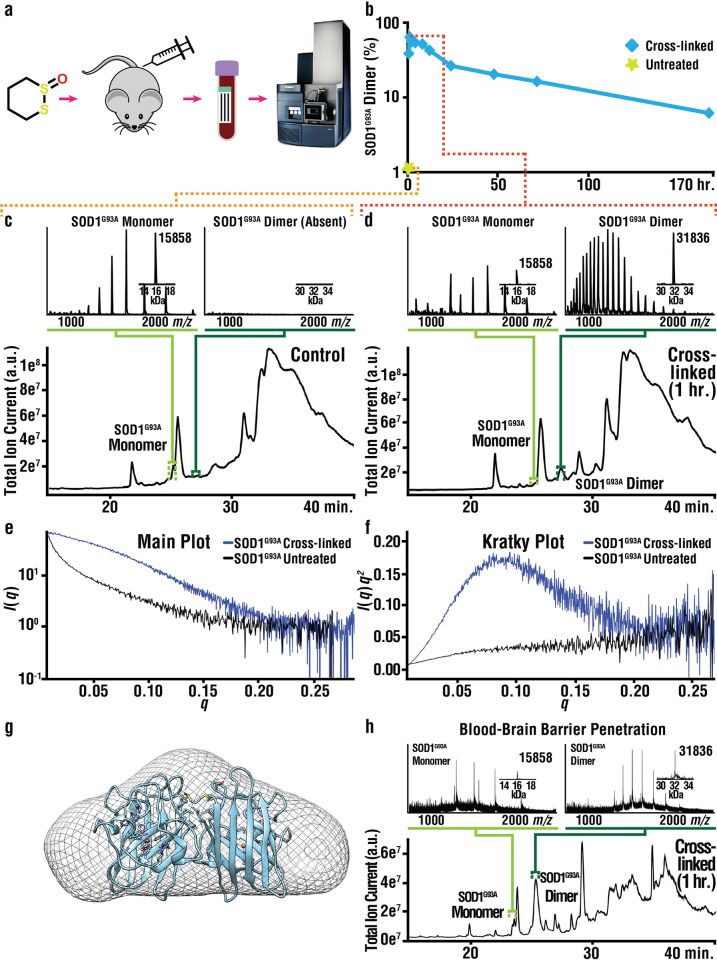
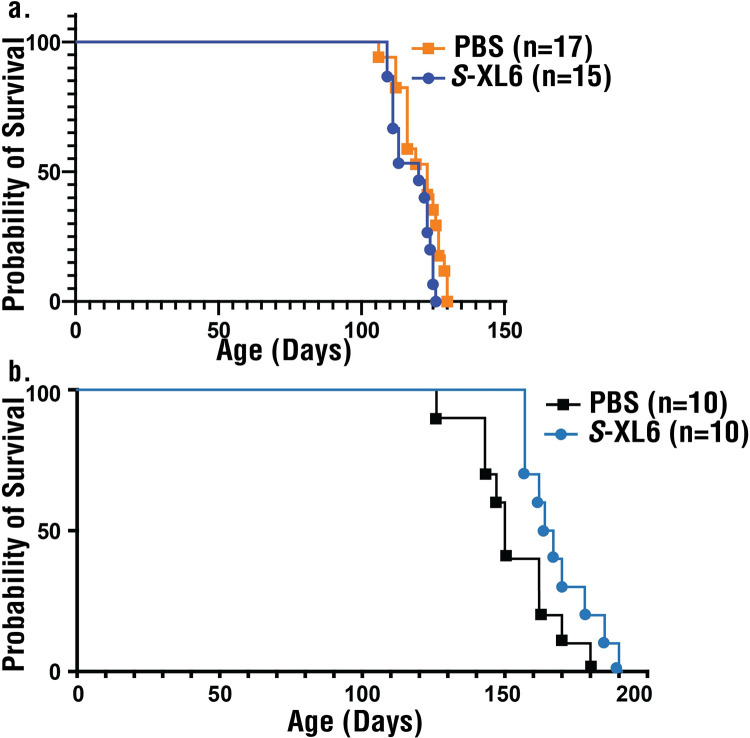
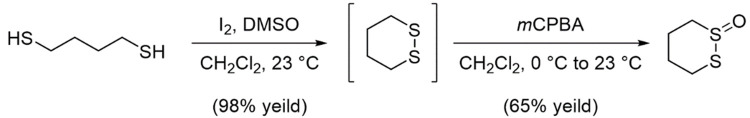
Mutations in the gene encoding Cu-Zn superoxide dismutase 1 (SOD1) cause a subset of familial amyotrophic lateral sclerosis (fALS) cases. A shared effect of these mutations is that SOD1, which is normally a stable dimer, dissociates into toxic monomers that seed toxic aggregates. Considerable research effort has been devoted to developing compounds that stabilize the dimer of fALS SOD1 variants, but unfortunately, this has not yet resulted in a treatment. We hypothesized that cyclic thiosulfinate cross-linkers, which selectively target a rare, 2 cysteine-containing motif, can stabilize fALS-causing SOD1 variants in vivo. We created a library of chemically diverse cyclic thiosulfinates and determined structure-cross-linking-activity relationships. A pre-lead compound, "S-XL6," was selected based upon its cross-linking rate and drug-like properties. Co-crystallographic structure clearly establishes the binding of S-XL6 at Cys 111 bridging the monomers and stabilizing the SOD1 dimer. Biophysical studies reveal that the degree of stabilization afforded by S-XL6 (up to 24°C) is unprecedented for fALS, and to our knowledge, for any protein target of any kinetic stabilizer. Gene silencing and protein degrading therapeutic approaches require careful dose titration to balance the benefit of diminished fALS SOD1 expression with the toxic loss-of-enzymatic function. We show that S-XL6 does not share this liability because it rescues the activity of fALS SOD1 variants. No pharmacological agent has been proven to bind to SOD1 in vivo. Here, using a fALS mouse model, we demonstrate oral bioavailability; rapid engagement of SOD1G93A by S-XL6 that increases SOD1G93A's in vivo half-life; and that S-XL6 crosses the blood-brain barrier. S-XL6 demonstrated a degree of selectivity by avoiding off-target binding to plasma proteins. Taken together, our results indicate that cyclic thiosulfinate-mediated SOD1 stabilization should receive further attention as a potential therapeutic approach for fALS.
Copyright: © 2024 Hossain et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
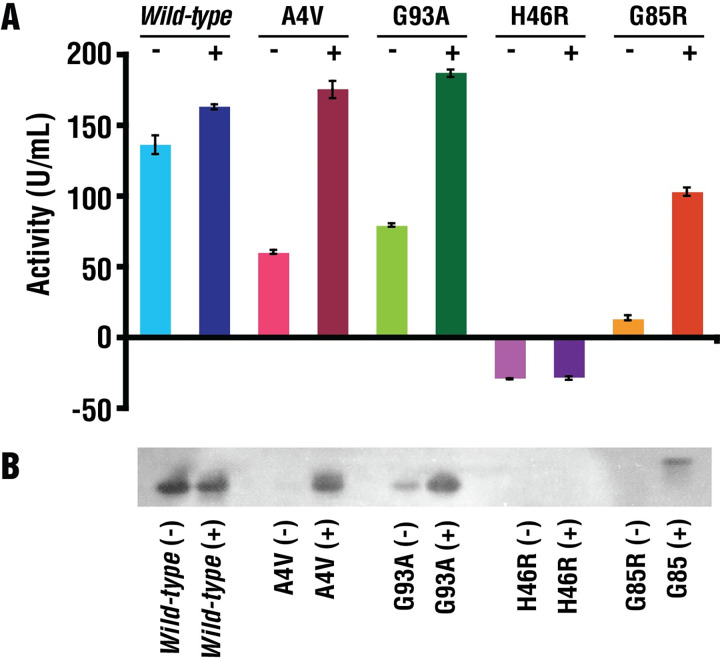
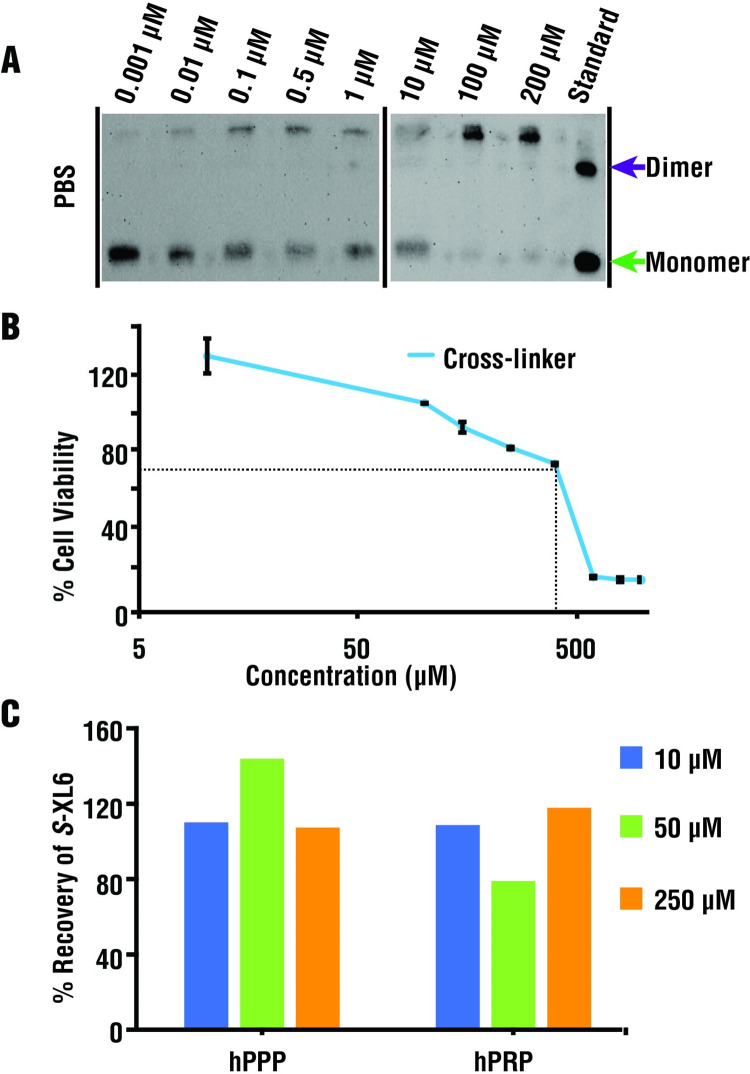
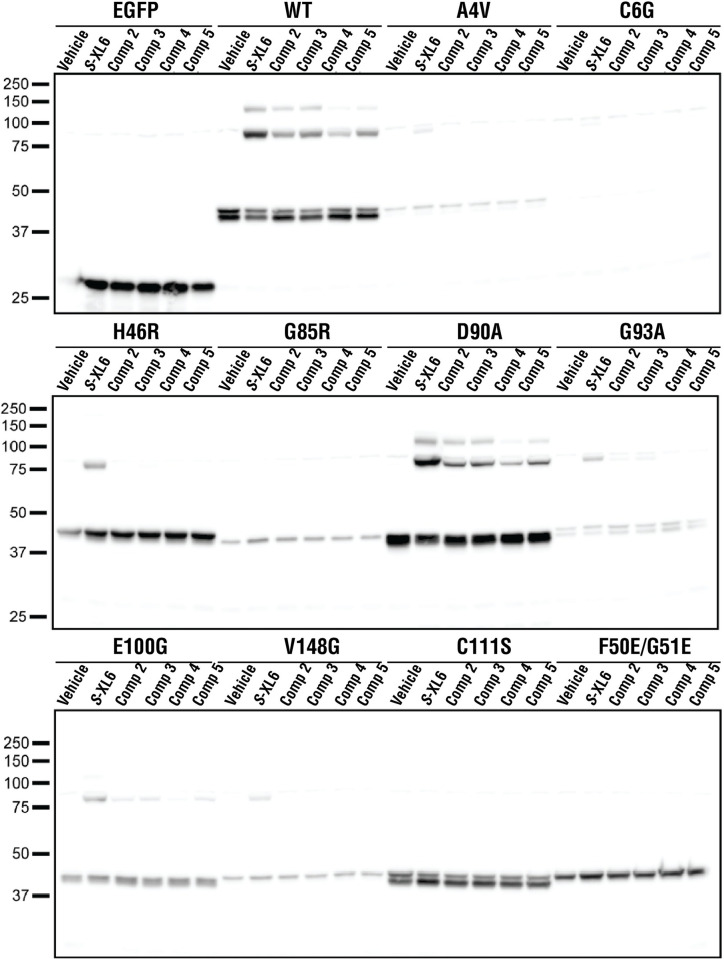
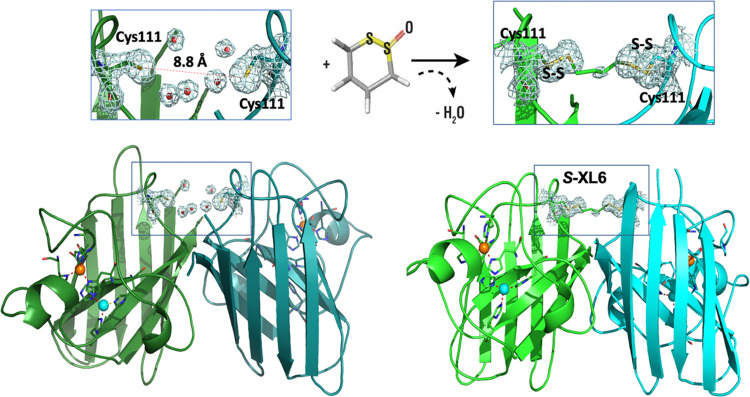
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
