

Cell-type-specific CAG repeat expansions and toxicity of mutant Huntingtin in human striatum and cerebellum
- PMID: 38291334
- PMCID: PMC10937393
- DOI: 10.1038/s41588-024-01653-6
Cell-type-specific CAG repeat expansions and toxicity of mutant Huntingtin in human striatum and cerebellum
Abstract
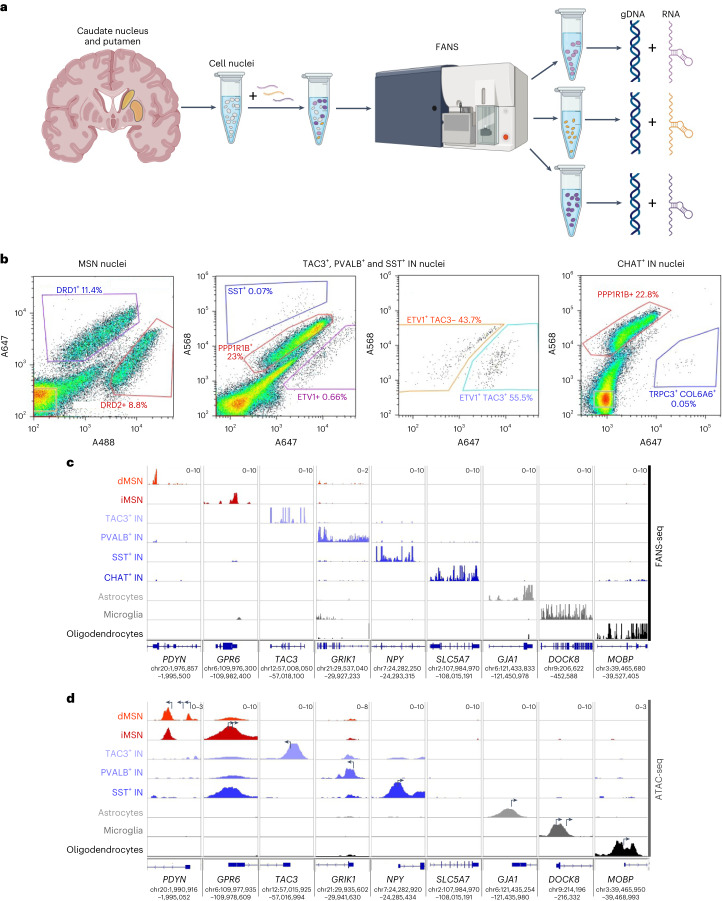
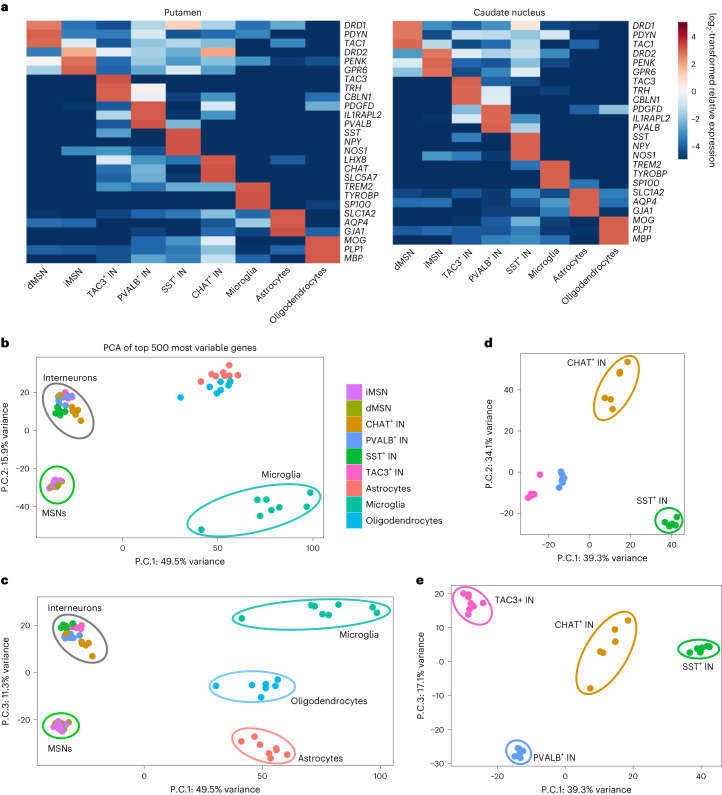
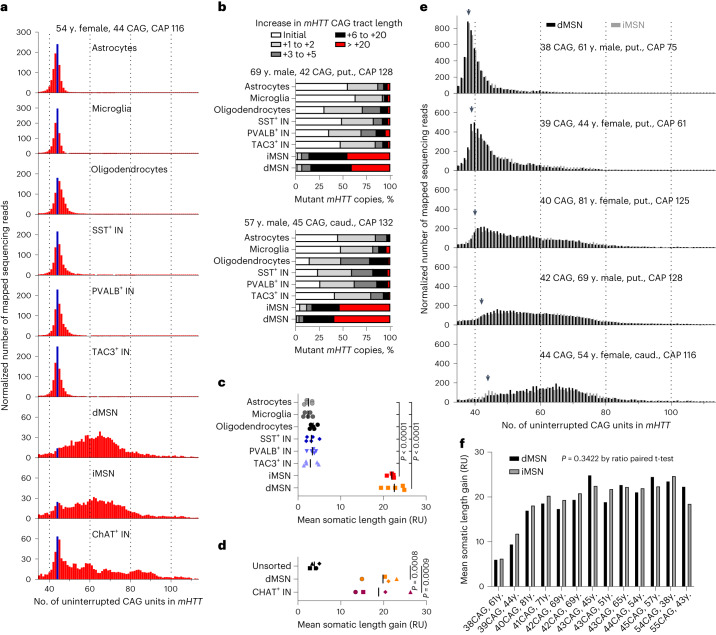
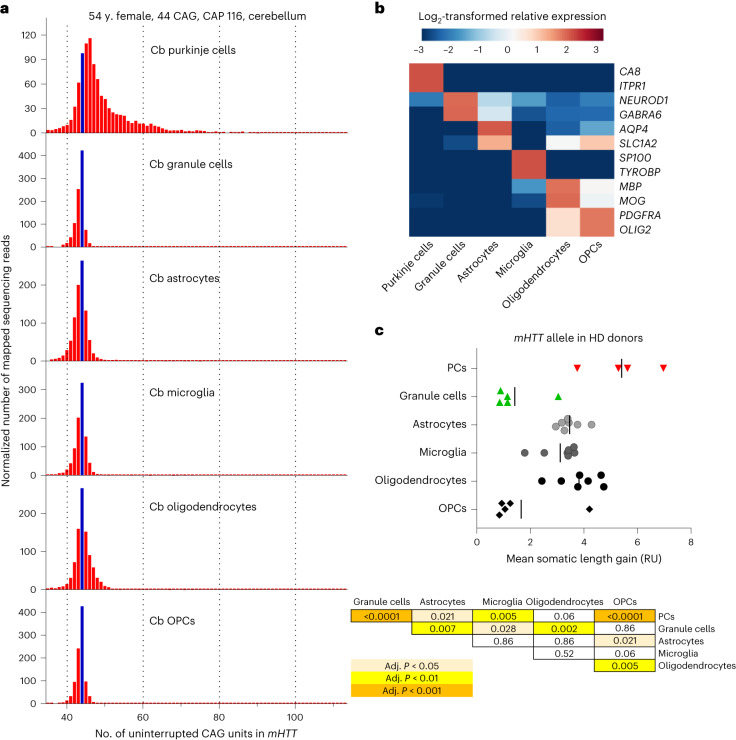
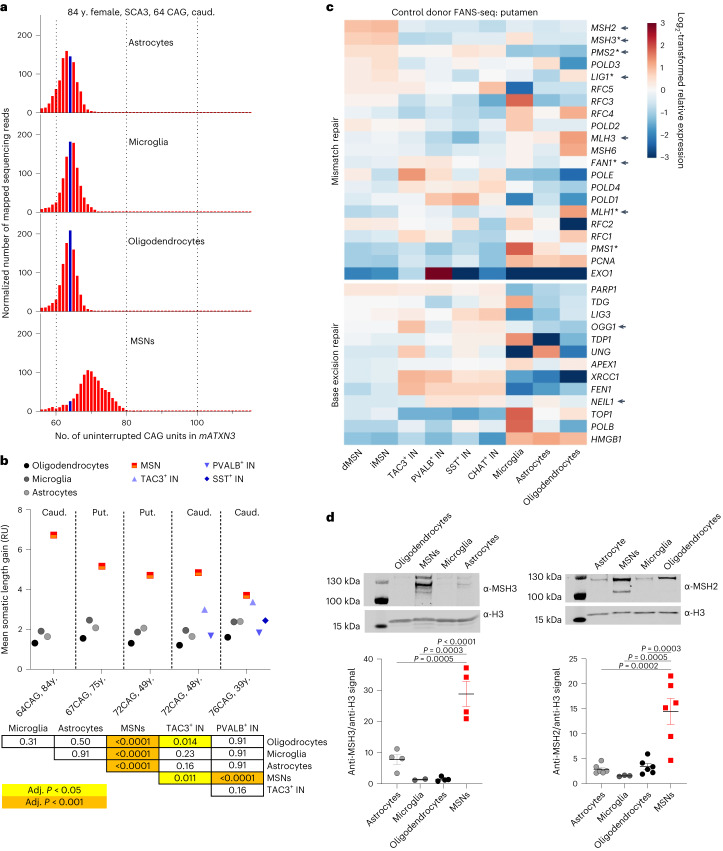
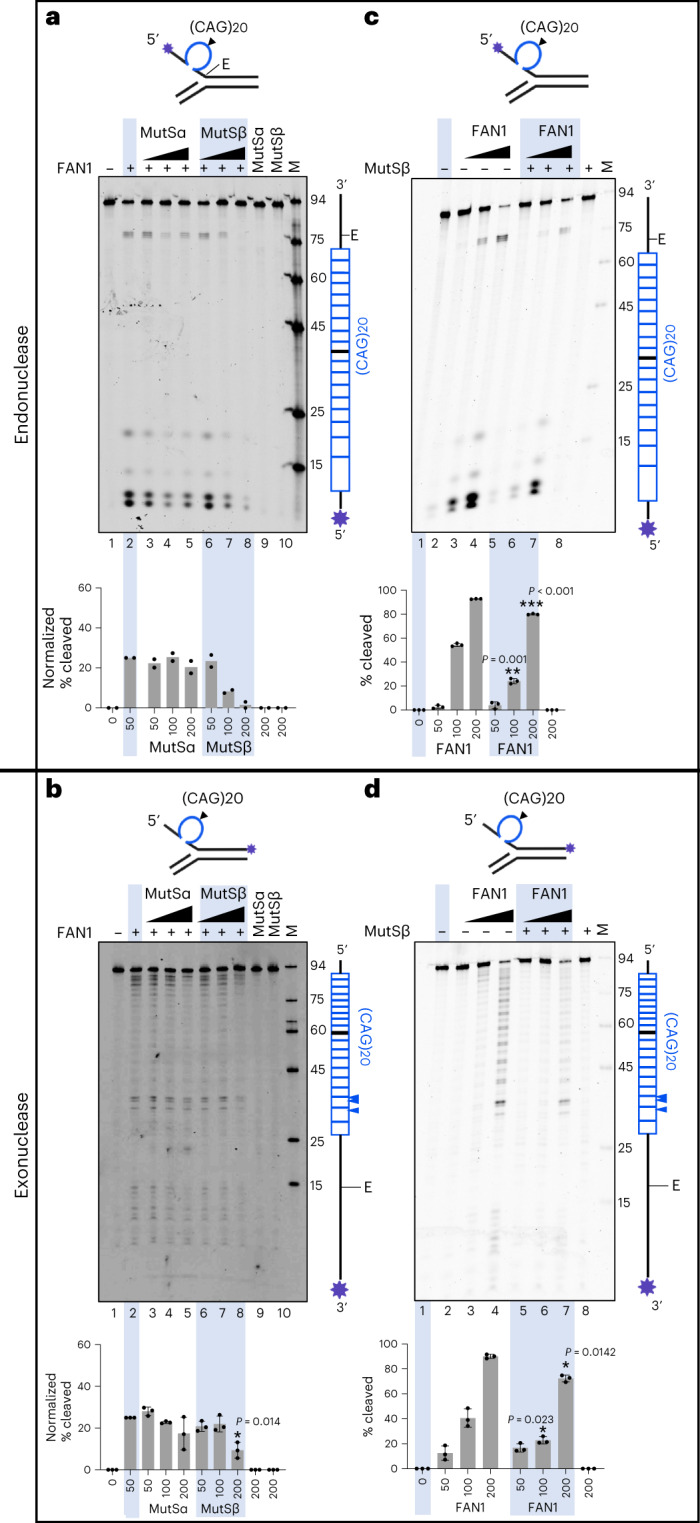
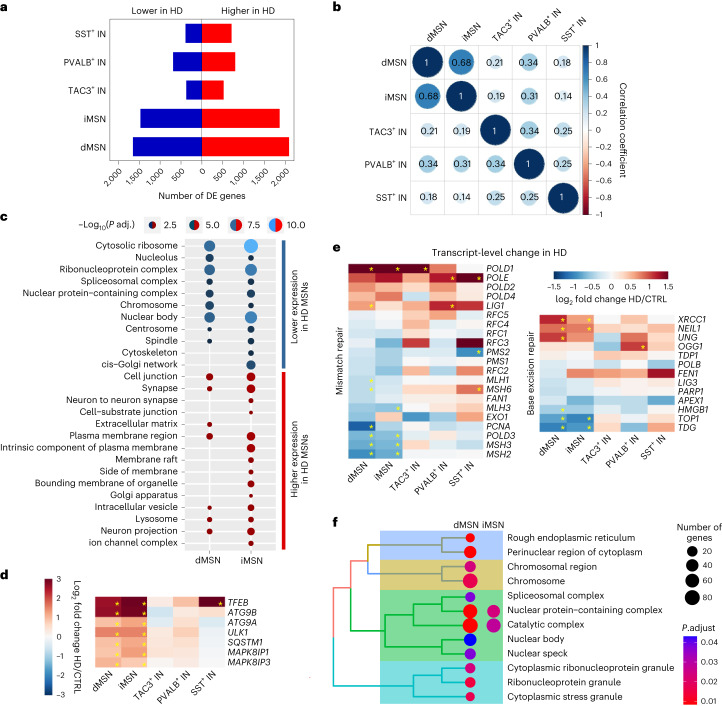
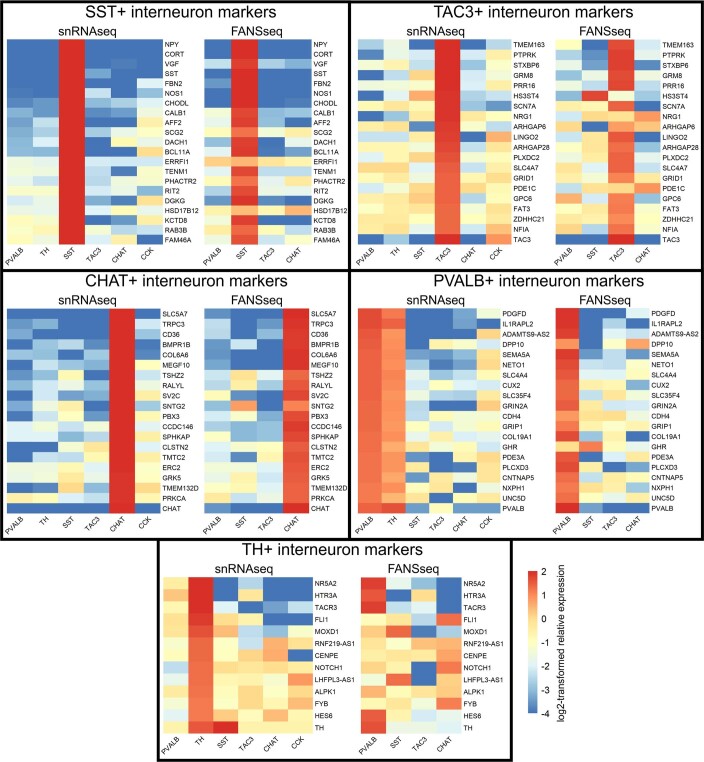
Brain region-specific degeneration and somatic expansions of the mutant Huntingtin (mHTT) CAG tract are key features of Huntington's disease (HD). However, the relationships among CAG expansions, death of specific cell types and molecular events associated with these processes are not established. Here, we used fluorescence-activated nuclear sorting (FANS) and deep molecular profiling to gain insight into the properties of cell types of the human striatum and cerebellum in HD and control donors. CAG expansions arise at mHTT in striatal medium spiny neurons (MSNs), cholinergic interneurons and cerebellar Purkinje neurons, and at mutant ATXN3 in MSNs from SCA3 donors. CAG expansions in MSNs are associated with higher levels of MSH2 and MSH3 (forming MutSβ), which can inhibit nucleolytic excision of CAG slip-outs by FAN1. Our data support a model in which CAG expansions are necessary but may not be sufficient for cell death and identify transcriptional changes associated with somatic CAG expansions and striatal toxicity.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Cell Type Specific CAG Repeat Expansions and Toxicity of Mutant Huntingtin in Human Striatum and Cerebellum.bioRxiv [Preprint]. 2023 Jun 8:2023.04.24.538082. doi: 10.1101/2023.04.24.538082. bioRxiv. 2023. Update in: Nat Genet. 2024 Mar;56(3):383-394. doi: 10.1038/s41588-024-01653-6. PMID: 37333326 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
