

This is a preprint.
Revealing protein sequence organization via contiguous hydrophobicity with the blobulator toolkit
- PMID: 38293114
- PMCID: PMC10827107
- DOI: 10.1101/2024.01.15.575761
Revealing protein sequence organization via contiguous hydrophobicity with the blobulator toolkit
Abstract
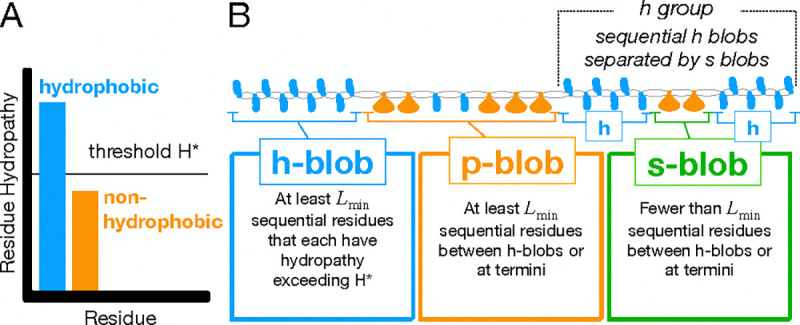
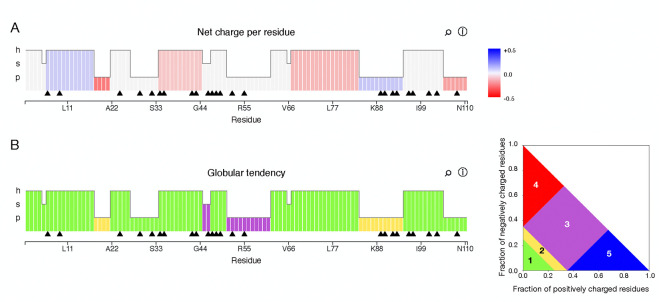
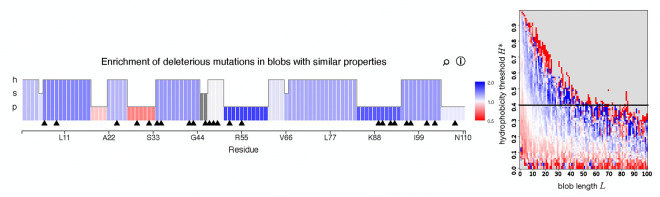
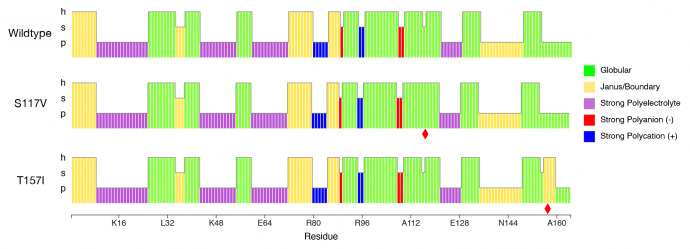
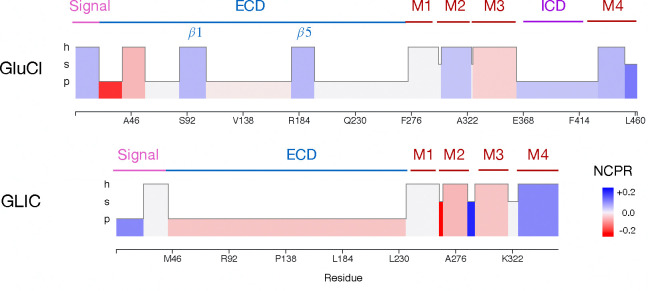
Clusters of hydrophobic residues are known to promote structured protein stability and drive protein aggregation. Recent work has shown that identifying contiguous hydrophobic residue clusters within protein sequences (termed "blobs") has proven useful in both intrinsically disordered protein (IDP) simulation and human genome studies. However, an accessible toolkit was unavailable, and the role that blobs play across the structural context of a variety of protein families remained unclear. Here, we present the blobulator toolkit: consisting of a webtool, a command line interface, and a VMD plugin. We demonstrate how identifying blobs using biologically relevant parameters provides useful information about a globular protein, two orthologous membrane proteins, and an IDP. Other potential applications are discussed, including: predicting protein segments with critical roles in tertiary interactions, providing a definition of local order and disorder with clear edges, and aiding in predicting protein features from sequence. The blobulator webtool can be found at www.blobulator.branniganlab.org, and the source code with pip installable command line tool, as well as the VMD plugin with installation instructions, can be found on GitHub at www.GitHub.com/BranniganLab/blobulator.
Figures









Similar articles
-
Proteus: a random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins.J Comput Aided Mol Des. 2017 May;31(5):453-466. doi: 10.1007/s10822-017-0020-y. Epub 2017 Apr 1. J Comput Aided Mol Des. 2017. PMID: 28365882 Free PMC article.
-
Epock: rapid analysis of protein pocket dynamics.Bioinformatics. 2015 May 1;31(9):1478-80. doi: 10.1093/bioinformatics/btu822. Epub 2014 Dec 12. Bioinformatics. 2015. PMID: 25505095 Free PMC article.
-
Contiguously hydrophobic sequences are functionally significant throughout the human exome.Proc Natl Acad Sci U S A. 2022 Mar 22;119(12):e2116267119. doi: 10.1073/pnas.2116267119. Epub 2022 Mar 16. Proc Natl Acad Sci U S A. 2022. PMID: 35294280 Free PMC article.
-
Intrinsically disordered proteins and multicellular organisms.Semin Cell Dev Biol. 2015 Jan;37:44-55. doi: 10.1016/j.semcdb.2014.09.025. Epub 2014 Oct 13. Semin Cell Dev Biol. 2015. PMID: 25307499 Review.
-
Order in Disorder as Observed by the "Hydrophobic Cluster Analysis" of Protein Sequences.Proteomics. 2018 Nov;18(21-22):e1800054. doi: 10.1002/pmic.201800054. Epub 2018 Oct 30. Proteomics. 2018. PMID: 30299594 Free PMC article. Review.
References
-
- Porollo A. A., Adamczak R., and Meller J., 2004. POLYVIEW: a flexible visualization tool for structural and functional annotations of proteins. Bioinformatics 20:2460–2462. - PubMed
-
- Porollo A., and Meller J., 2007. Prediction-based fingerprints of protein–protein interactions. PROTEINS: Structure, Function, and Bioinformatics . - PubMed
-
- Deleage G., Combet C., Blanchet C., and Geourjon C., 2001. ANTHEPROT: An integrated protein sequence analysis software with client/server capabilities. Computers in Biology and Medicine . - PubMed
-
- Dyson H. J., and Wright P. E., 2002. Coupling of folding and binding for unstructured proteins. Current Opinion in Structural Biology . - PubMed
-
- Ward J., Sodhi J., McGuffin L., Buxton B., and Jones D., 2004. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. Journal of Molecular Biology . - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources