

This is a preprint.
Gamma protocadherins in vascular endothelial cells inhibit Klf2/4 to promote atherosclerosis
- PMID: 38293157
- PMCID: PMC10827163
- DOI: 10.1101/2024.01.16.575958
Gamma protocadherins in vascular endothelial cells inhibit Klf2/4 to promote atherosclerosis
Update in
-
Endothelial γ-protocadherins inhibit KLF2 and KLF4 to promote atherosclerosis.Nat Cardiovasc Res. 2024 Sep;3(9):1035-1048. doi: 10.1038/s44161-024-00522-z. Epub 2024 Sep 4. Nat Cardiovasc Res. 2024. PMID: 39232138 Free PMC article.
Abstract
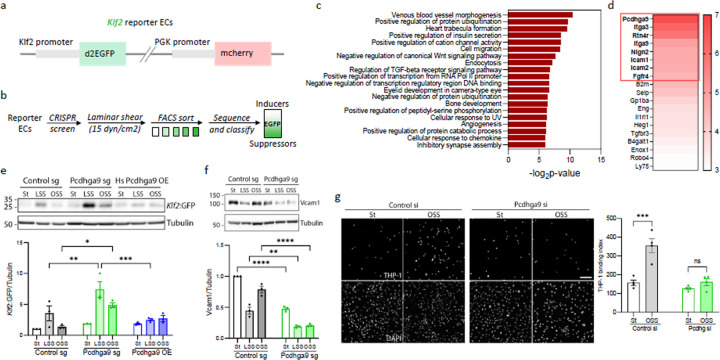
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of mortality worldwide1. Laminar shear stress (LSS) from blood flow in straight regions of arteries protects against ASCVD by upregulating the Klf2/4 anti-inflammatory program in endothelial cells (ECs)2-8. Conversely, disturbed shear stress (DSS) at curves or branches predisposes these regions to plaque formation9,10. We previously reported a whole genome CRISPR knockout screen11 that identified novel inducers of Klf2/4. Here we report suppressors of Klf2/4 and characterize one candidate, protocadherin gamma A9 (Pcdhga9), a member of the clustered protocadherin gene family12. Pcdhg deletion increases Klf2/4 levels in vitro and in vivo and suppresses inflammatory activation of ECs. Pcdhg suppresses Klf2/4 by inhibiting the Notch pathway via physical interaction of cleaved Notch1 intracellular domain (NICD Val1744) with nuclear Pcdhg C-terminal constant domain (CCD). Pcdhg inhibition by EC knockout (KO) or blocking antibody protects from atherosclerosis. Pcdhg is elevated in the arteries of human atherosclerosis. This study identifies a novel fundamental mechanism of EC resilience and therapeutic target for treating inflammatory vascular disease.
Keywords: fluid shear stress; mechanotransduction; protocadherin gamma A9; vascular inflammation.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials