

Systems immunology of transcriptional responses to viral infection identifies conserved antiviral pathways across macaques and humans
- PMID: 38294906
- PMCID: PMC10915397
- DOI: 10.1016/j.celrep.2024.113706
Systems immunology of transcriptional responses to viral infection identifies conserved antiviral pathways across macaques and humans
Abstract
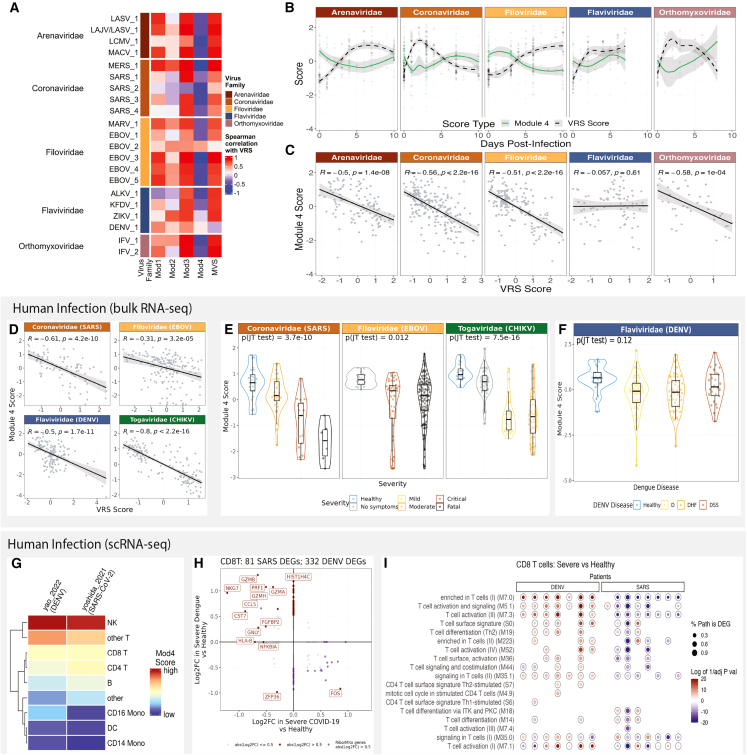
Viral pandemics and epidemics pose a significant global threat. While macaque models of viral disease are routinely used, it remains unclear how conserved antiviral responses are between macaques and humans. Therefore, we conducted a cross-species analysis of transcriptomic data from over 6,088 blood samples from macaques and humans infected with one of 31 viruses. Our findings demonstrate that irrespective of primate or viral species, there are conserved antiviral responses that are consistent across infection phase (acute, chronic, or latent) and viral genome type (DNA or RNA viruses). Leveraging longitudinal data from experimental challenges, we identify virus-specific response kinetics such as host responses to Coronaviridae and Orthomyxoviridae infections peaking 1-3 days earlier than responses to Filoviridae and Arenaviridae viral infections. Our results underscore macaque studies as a powerful tool for understanding viral pathogenesis and immune responses that translate to humans, with implications for viral therapeutic development and pandemic preparedness.
Keywords: CP: Immunology; antiviral immunity; conserved host response to viruses; human immune response; macaques; non-human primates; systems immunology; transcriptomics; virus.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures





References
-
- Domingo E., Holland J.J. RNA VIRUS MUTATIONS AND FITNESS FOR SURVIVAL. Annu. Rev. Microbiol. 1997;51:151–178. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
