

Folate depletion induces erythroid differentiation through perturbation of de novo purine synthesis
- PMID: 38295180
- PMCID: PMC10830111
- DOI: 10.1126/sciadv.adj9479
Folate depletion induces erythroid differentiation through perturbation of de novo purine synthesis
Abstract
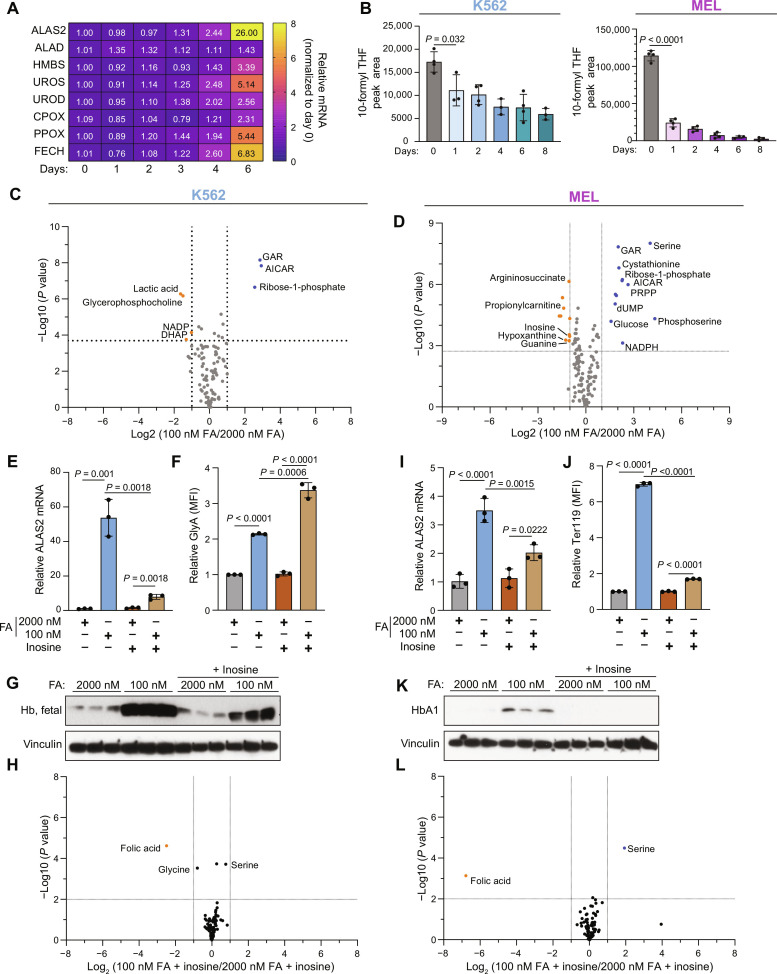
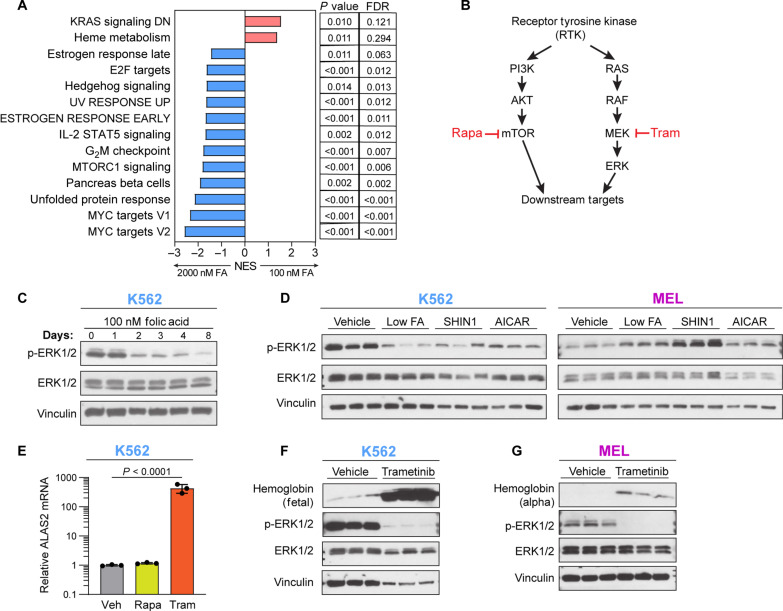
Folate, an essential vitamin, is a one-carbon acceptor and donor in key metabolic reactions. Erythroid cells harbor a unique sensitivity to folate deprivation, as revealed by the primary pathological manifestation of nutritional folate deprivation: megaloblastic anemia. To study this metabolic sensitivity, we applied mild folate depletion to human and mouse erythroid cell lines and primary murine erythroid progenitors. We show that folate depletion induces early blockade of purine synthesis and accumulation of the purine synthesis intermediate and signaling molecule, 5'-phosphoribosyl-5-aminoimidazole-4-carboxamide (AICAR), followed by enhanced heme metabolism, hemoglobin synthesis, and erythroid differentiation. This is phenocopied by inhibition of folate metabolism using the inhibitor SHIN1, and by AICAR supplementation. Mechanistically, the metabolically driven differentiation is independent of mechanistic target of rapamycin complex 1 (mTORC1) and adenosine 5'-monophosphate-activated protein kinase (AMPK) and is instead mediated by protein kinase C. Our findings suggest that folate deprivation-induced premature differentiation of erythroid progenitor cells is a molecular etiology to folate deficiency-induced anemia.
Figures






References
-
- Hitchings G. H., Burchall J. J., Inhibition of folate biosynthesis and function as a basis for chemotherapy. Adv. Enzymol. Relat. Areas Mol. Biol. 27, 417–468 (1965). - PubMed
-
- Wilkinson J. F., Israels M. C., Fletcher F., Folic acid in the treatment of pernicious anaemia. Lancet 2, 156–158 (1946). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
