

Stress response silencing by an E3 ligase mutated in neurodegeneration
- PMID: 38297121
- PMCID: PMC10881396
- DOI: 10.1038/s41586-023-06985-7
Stress response silencing by an E3 ligase mutated in neurodegeneration
Abstract
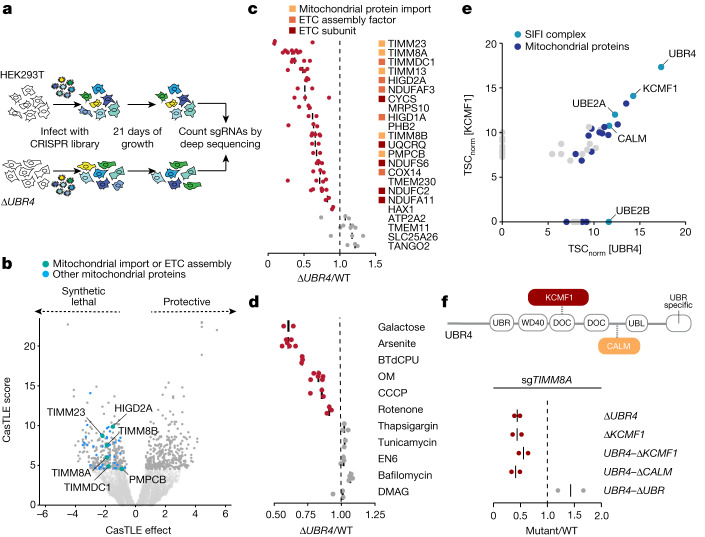
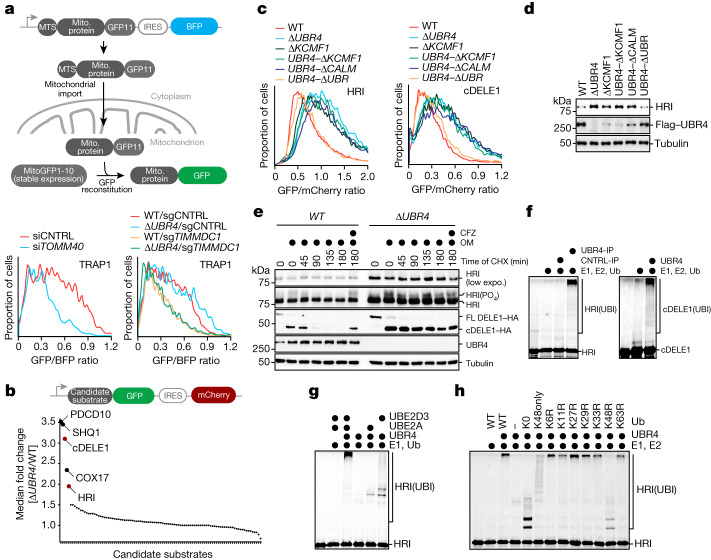
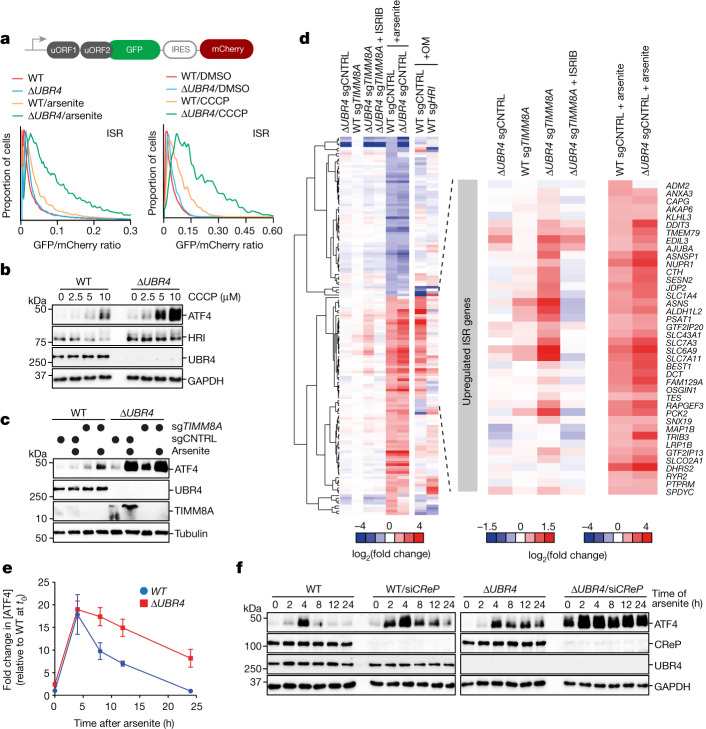
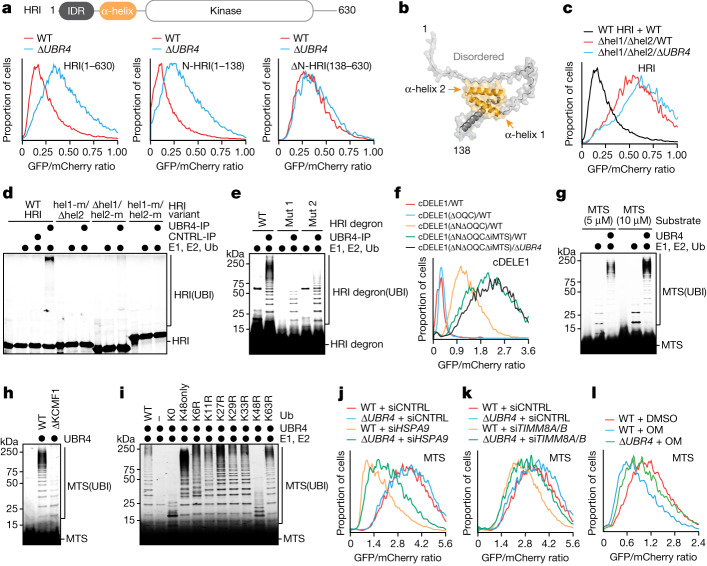
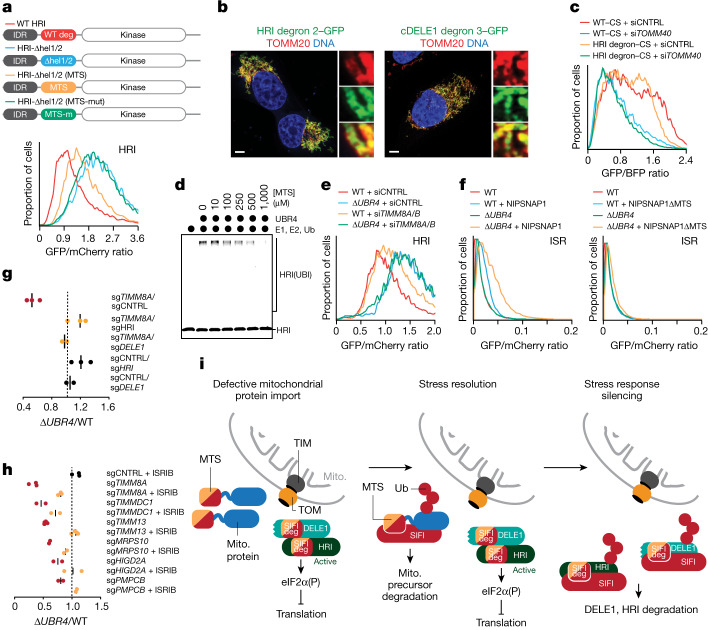
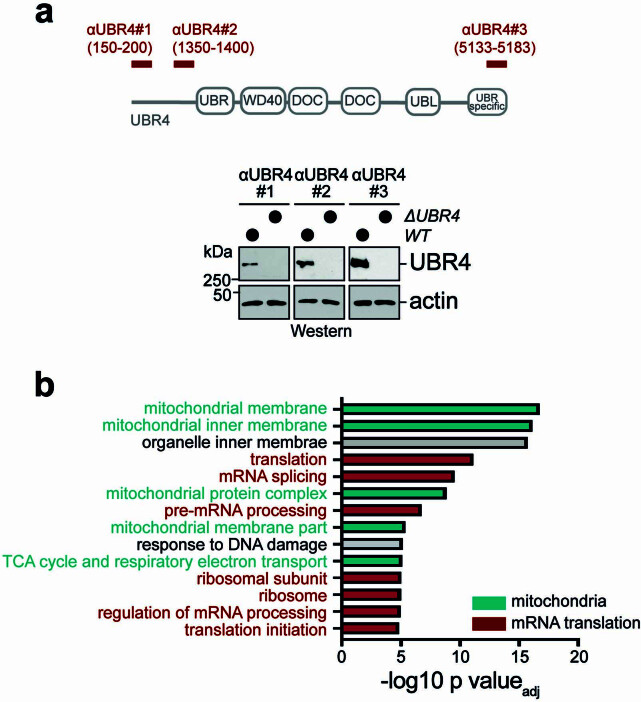
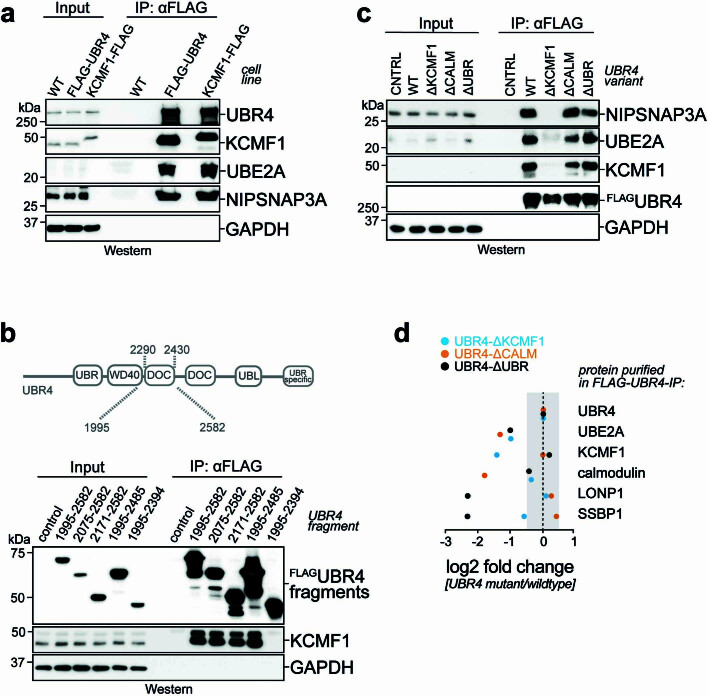
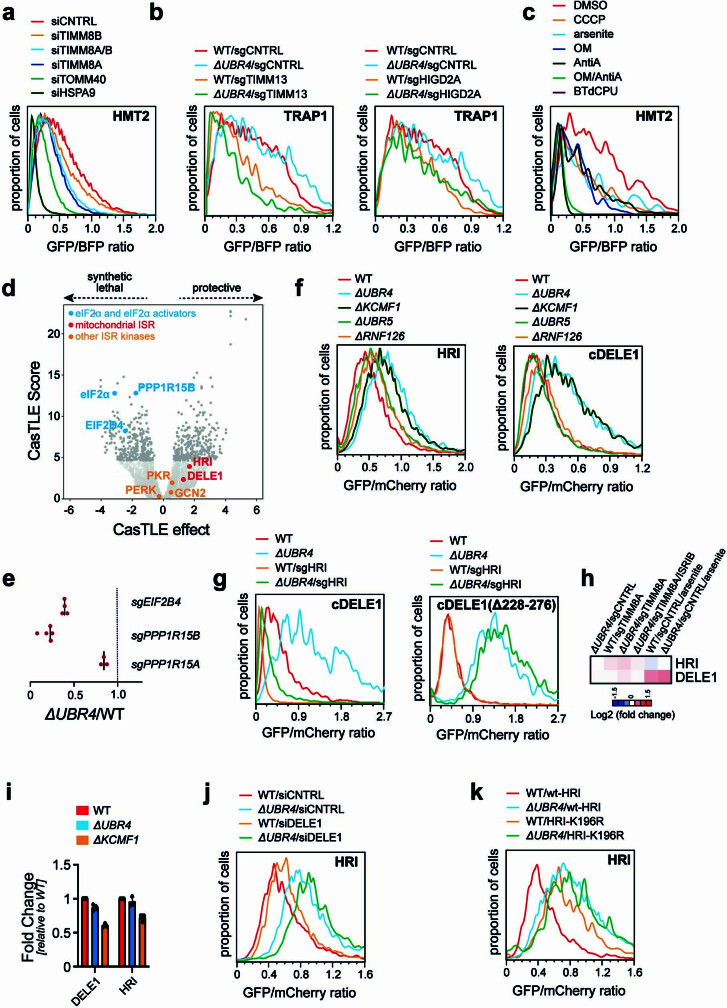
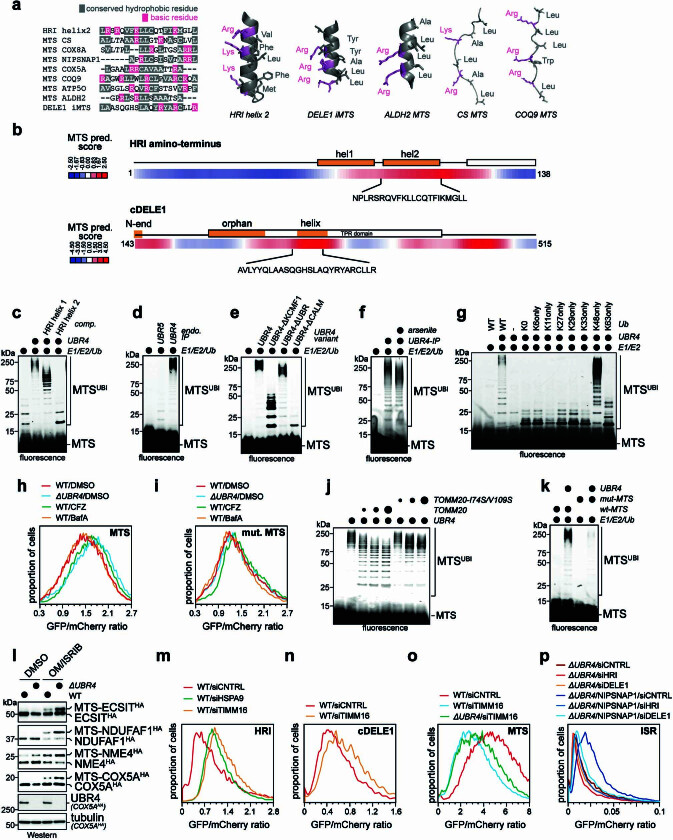
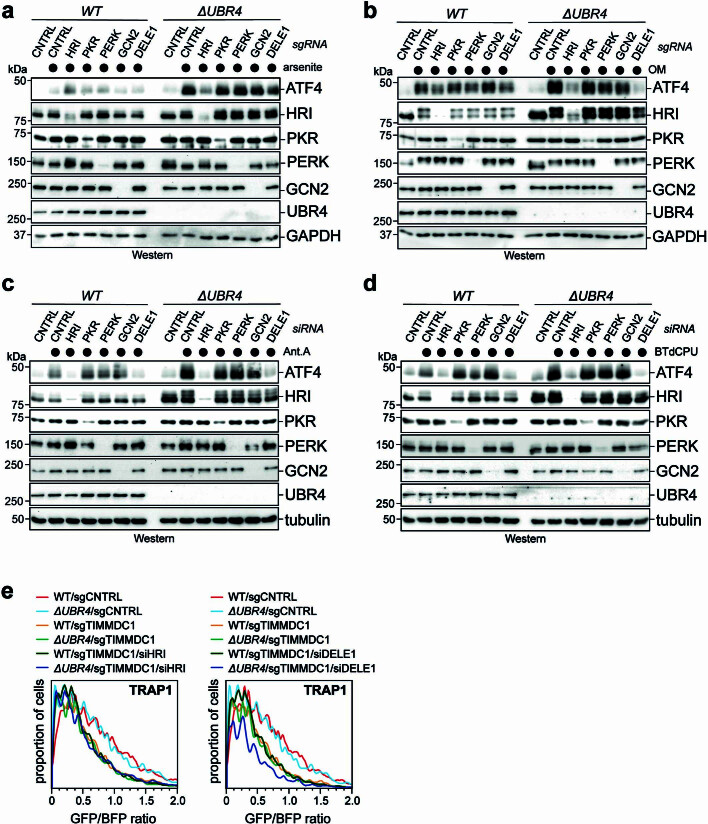
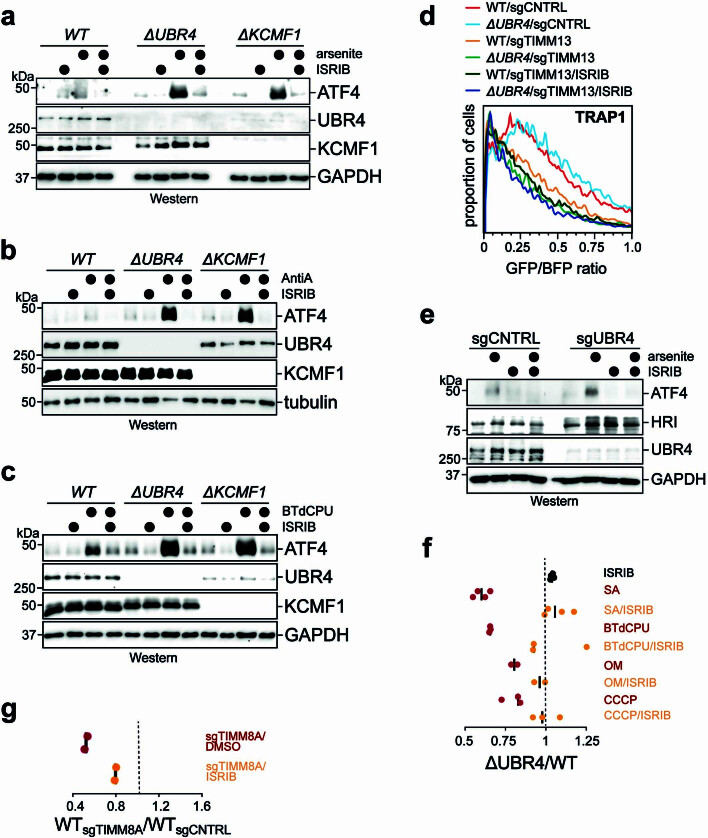
Stress response pathways detect and alleviate adverse conditions to safeguard cell and tissue homeostasis, yet their prolonged activation induces apoptosis and disrupts organismal health1-3. How stress responses are turned off at the right time and place remains poorly understood. Here we report a ubiquitin-dependent mechanism that silences the cellular response to mitochondrial protein import stress. Crucial to this process is the silencing factor of the integrated stress response (SIFI), a large E3 ligase complex mutated in ataxia and in early-onset dementia that degrades both unimported mitochondrial precursors and stress response components. By recognizing bifunctional substrate motifs that equally encode protein localization and stability, the SIFI complex turns off a general stress response after a specific stress event has been resolved. Pharmacological stress response silencing sustains cell survival even if stress resolution failed, which underscores the importance of signal termination and provides a roadmap for treating neurodegenerative diseases caused by mitochondrial import defects.
© 2024. The Author(s).
Conflict of interest statement
M.R. is co-founder and SAB member of Nurix Therapeutics, Zenith Therapeutics and Lyterian Therapeutics, SAB member of Vicinitas Therapeutics, and an iPartner of The Column Group Ventures. M.W. is co-founder and SAB member of Lyterian Therapeutics. All other authors declare no conflicts of interest.
Figures
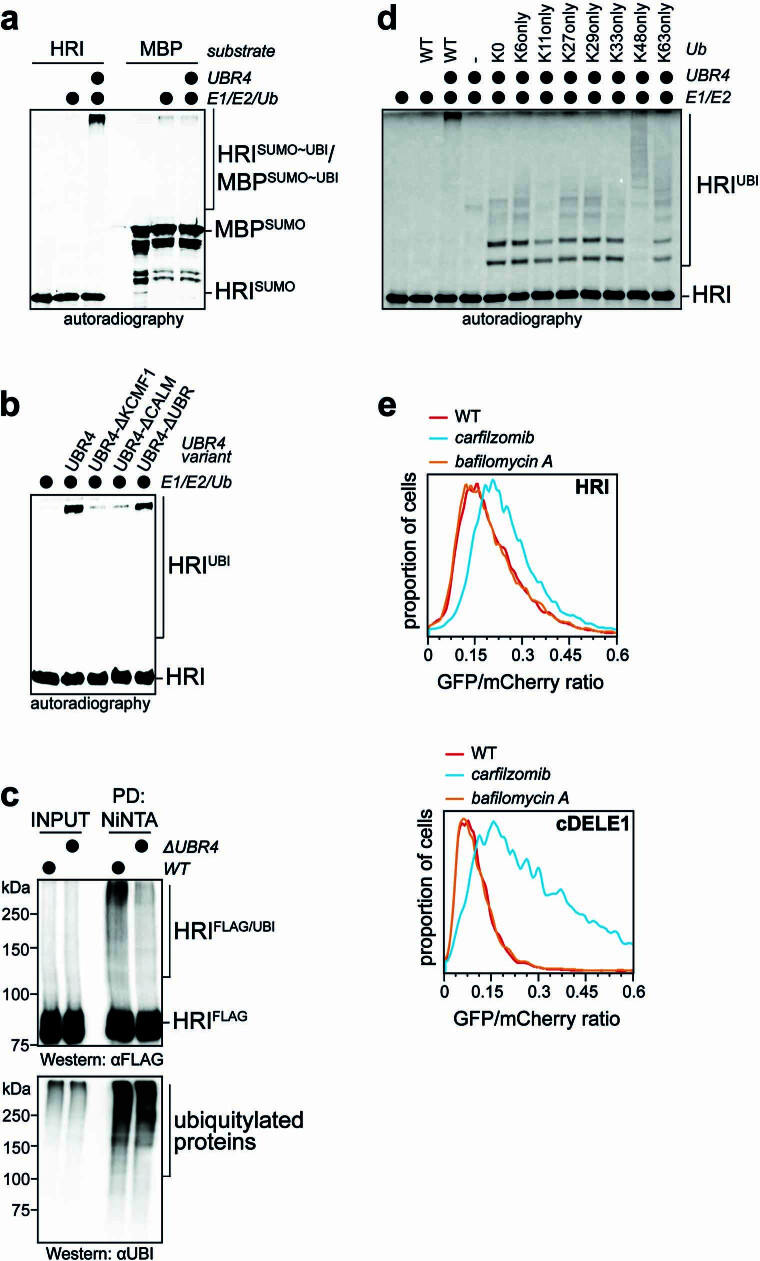
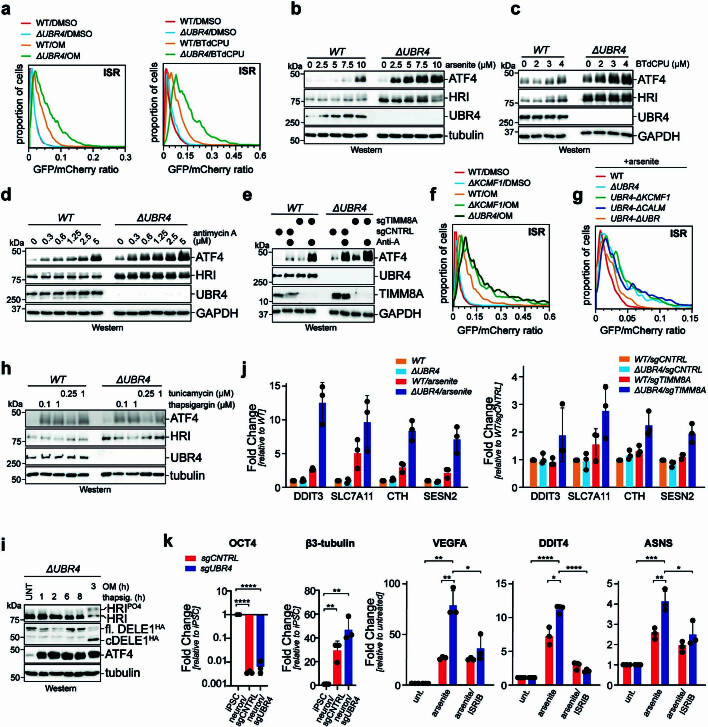
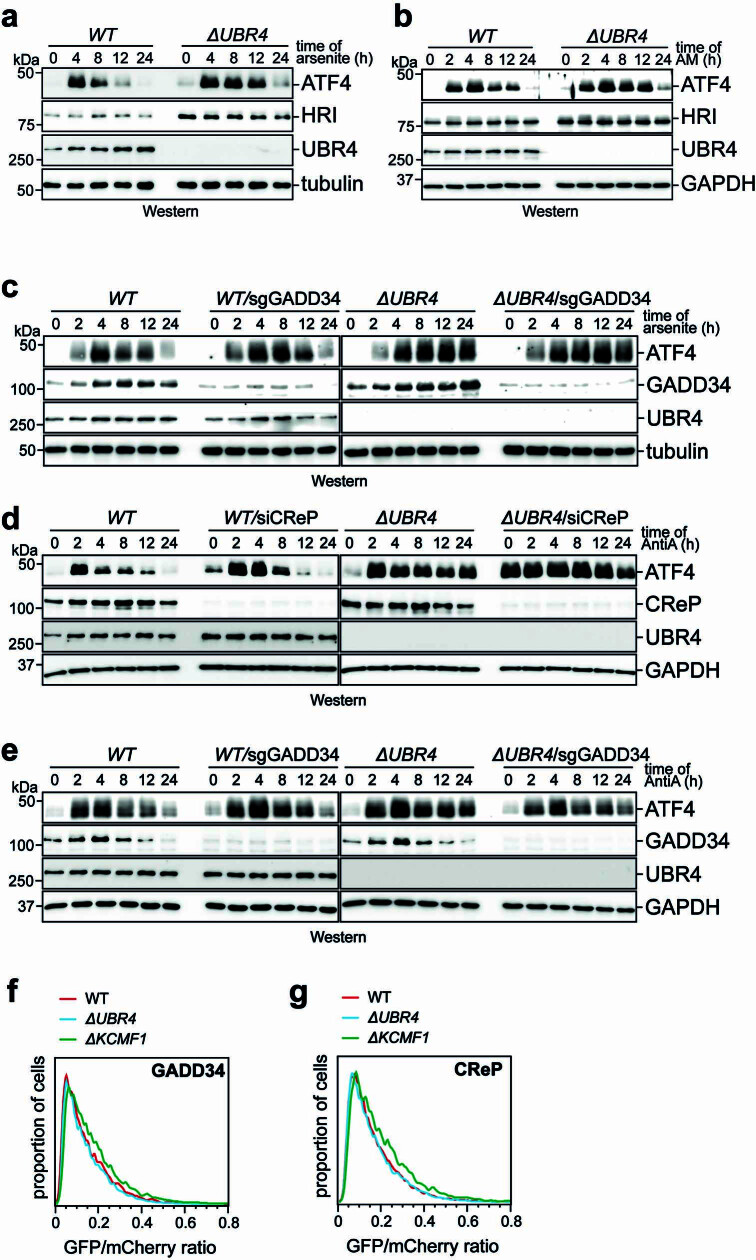
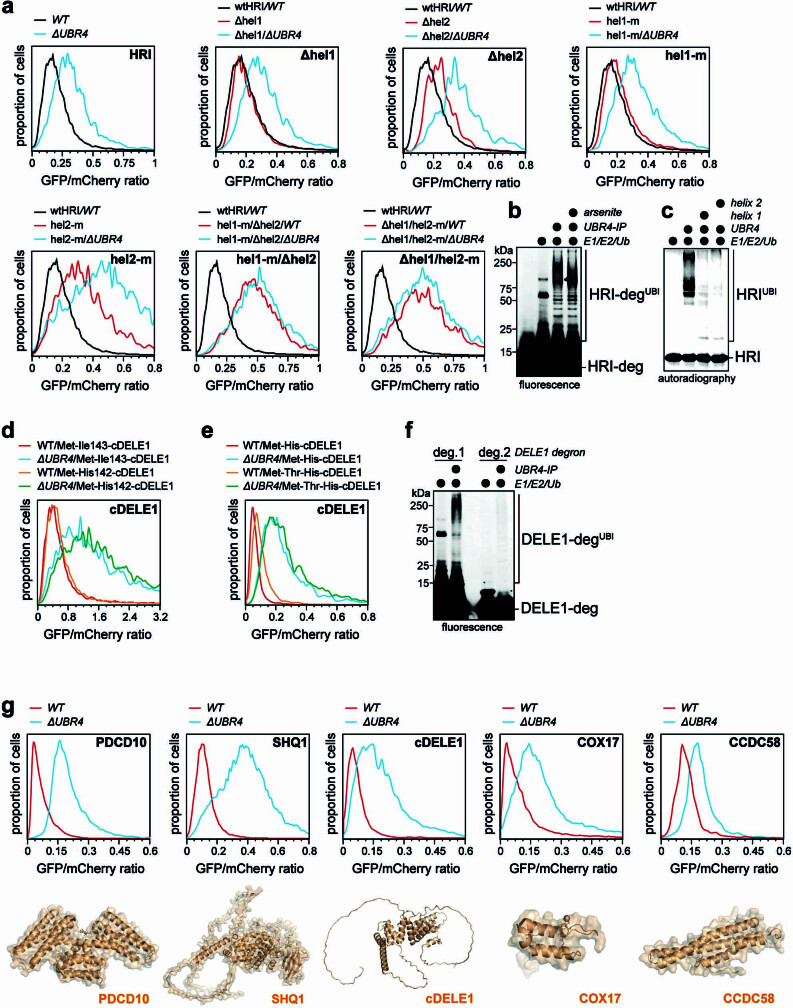
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
