

Stereodivergent 1,3-difunctionalization of alkenes by charge relocation
- PMID: 38297174
- PMCID: PMC10830407
- DOI: 10.1038/s41586-023-06938-0
Stereodivergent 1,3-difunctionalization of alkenes by charge relocation
Abstract
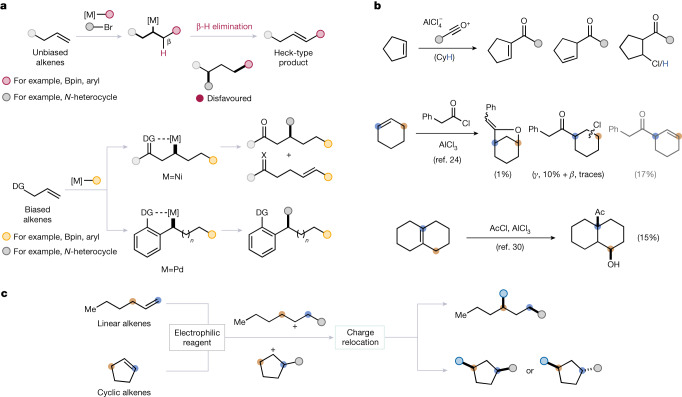
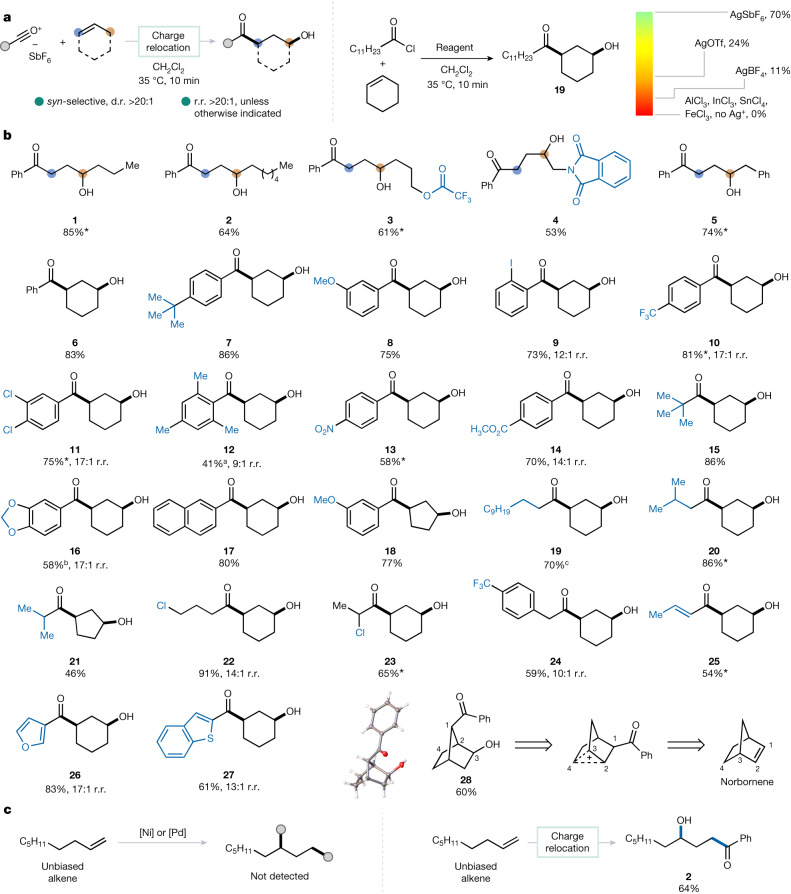
Alkenes are indispensable feedstocks in chemistry. Functionalization at both carbons of the alkene-1,2-difunctionalization-is part of chemistry curricula worldwide1. Although difunctionalization at distal positions has been reported2-4, it typically relies on designer substrates featuring directing groups and/or stabilizing features, all of which determine the ultimate site of bond formation5-7. Here we introduce a method for the direct 1,3-difunctionalization of alkenes, based on a concept termed 'charge relocation', which enables stereodivergent access to 1,3-difunctionalized products of either syn- or anti-configuration from unactivated alkenes, without the need for directing groups or stabilizing features. The usefulness of the approach is demonstrated in the synthesis of the pulmonary toxin 4-ipomeanol and its derivatives.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Clayden, J., Greeves, N. & Warren S. Organic Chemistry (Oxford Univ. Press, 2012).
-
- Massad I, et al. Stereoselective synthesis through remote functionalization. Nat. Synth. 2022;1:37–48. doi: 10.1038/s44160-021-00002-3. - DOI
-
- Wang, W. et al. Migratory arylboration of unactivated alkenes enabled by nickel catalysis. Angew. Chem. Int. Ed.58, 4612–4616 (2019). - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
