

Heterozygous loss-of-function SMC3 variants are associated with variable growth and developmental features
- PMID: 38297832
- PMCID: PMC10876629
- DOI: 10.1016/j.xhgg.2024.100273
Heterozygous loss-of-function SMC3 variants are associated with variable growth and developmental features
Abstract
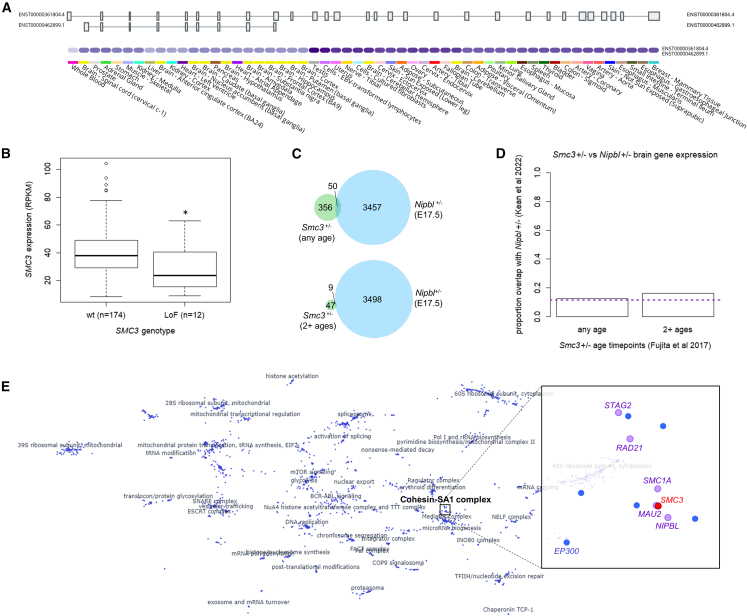
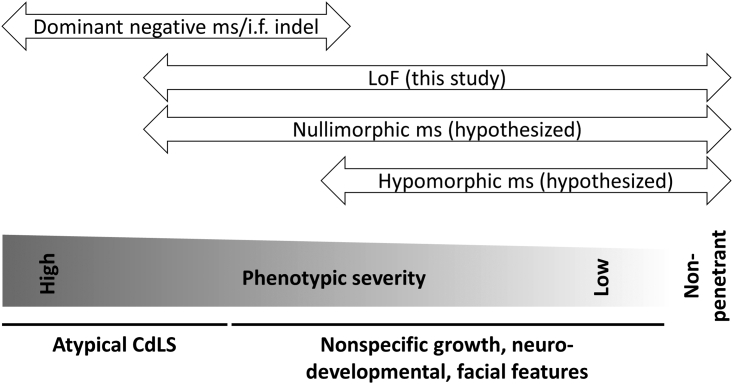
Heterozygous missense variants and in-frame indels in SMC3 are a cause of Cornelia de Lange syndrome (CdLS), marked by intellectual disability, growth deficiency, and dysmorphism, via an apparent dominant-negative mechanism. However, the spectrum of manifestations associated with SMC3 loss-of-function variants has not been reported, leading to hypotheses of alternative phenotypes or even developmental lethality. We used matchmaking servers, patient registries, and other resources to identify individuals with heterozygous, predicted loss-of-function (pLoF) variants in SMC3, and analyzed population databases to characterize mutational intolerance in this gene. Here, we show that SMC3 behaves as an archetypal haploinsufficient gene: it is highly constrained against pLoF variants, strongly depleted for missense variants, and pLoF variants are associated with a range of developmental phenotypes. Among 14 individuals with SMC3 pLoF variants, phenotypes were variable but coalesced on low growth parameters, developmental delay/intellectual disability, and dysmorphism, reminiscent of atypical CdLS. Comparisons to individuals with SMC3 missense/in-frame indel variants demonstrated an overall milder presentation in pLoF carriers. Furthermore, several individuals harboring pLoF variants in SMC3 were nonpenetrant for growth, developmental, and/or dysmorphic features, and some had alternative symptomatologies with rational biological links to SMC3. Analyses of tumor and model system transcriptomic data and epigenetic data in a subset of cases suggest that SMC3 pLoF variants reduce SMC3 expression but do not strongly support clustering with functional genomic signatures of typical CdLS. Our finding of substantial population-scale LoF intolerance in concert with variable growth and developmental features in subjects with SMC3 pLoF variants expands the scope of cohesinopathies, informs on their allelic architecture, and suggests the existence of additional clearly LoF-constrained genes whose disease links will be confirmed only by multilayered genomic data paired with careful phenotyping.
Keywords: CdLS3; Cornelia de Lange syndrome; LoF; SMC3; cohesin; loss-of-function.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.E.T. is supported by research funding and/or reagents from Illumina, Microsoft, Ionis Therapeutics, and Levo Therapeutics. M.P.N., J.H., and J.J. are employees of GeneDx.
Figures



Update of
-
Heterozygous loss-of-function SMC3 variants are associated with variable and incompletely penetrant growth and developmental features.medRxiv [Preprint]. 2023 Sep 28:2023.09.27.23294269. doi: 10.1101/2023.09.27.23294269. medRxiv. 2023. Update in: HGG Adv. 2024 Apr 11;5(2):100273. doi: 10.1016/j.xhgg.2024.100273. PMID: 37808847 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- P50 HD105351/HD/NICHD NIH HHS/United States
- R01 HD078641/HD/NICHD NIH HHS/United States
- R01 HG009141/HG/NHGRI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- K23 NS119666/NS/NINDS NIH HHS/United States
- K08 NS117891/NS/NINDS NIH HHS/United States
- R01 EY012910/EY/NEI NIH HHS/United States
- U01 HG011755/HG/NHGRI NIH HHS/United States
- P30 EY014104/EY/NEI NIH HHS/United States
- MC_UU_00007/3/MRC_/Medical Research Council/United Kingdom
- R01 HL143295/HL/NHLBI NIH HHS/United States
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- R01 EY026904/EY/NEI NIH HHS/United States
- P50 HD104224/HD/NICHD NIH HHS/United States
- R01 MH115957/MH/NIMH NIH HHS/United States
- R01 HD081256/HD/NICHD NIH HHS/United States
- P50 HD096723/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
