

CSN6-SPOP-HMGCS1 Axis Promotes Hepatocellular Carcinoma Progression via YAP1 Activation
- PMID: 38308184
- PMCID: PMC11005689
- DOI: 10.1002/advs.202306827
CSN6-SPOP-HMGCS1 Axis Promotes Hepatocellular Carcinoma Progression via YAP1 Activation
Abstract
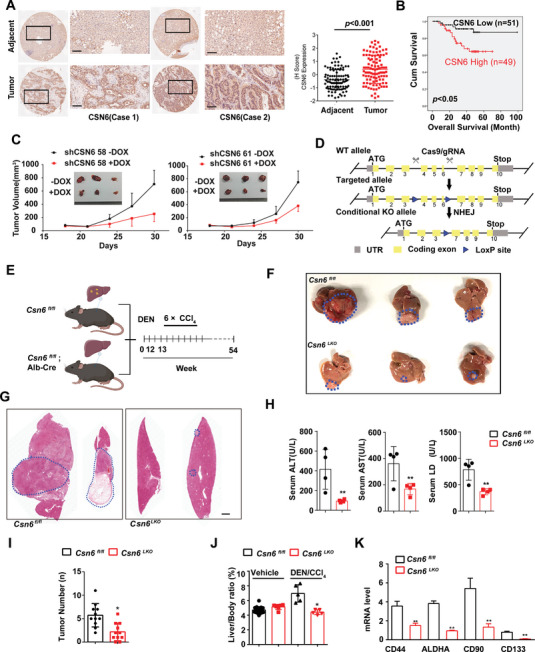
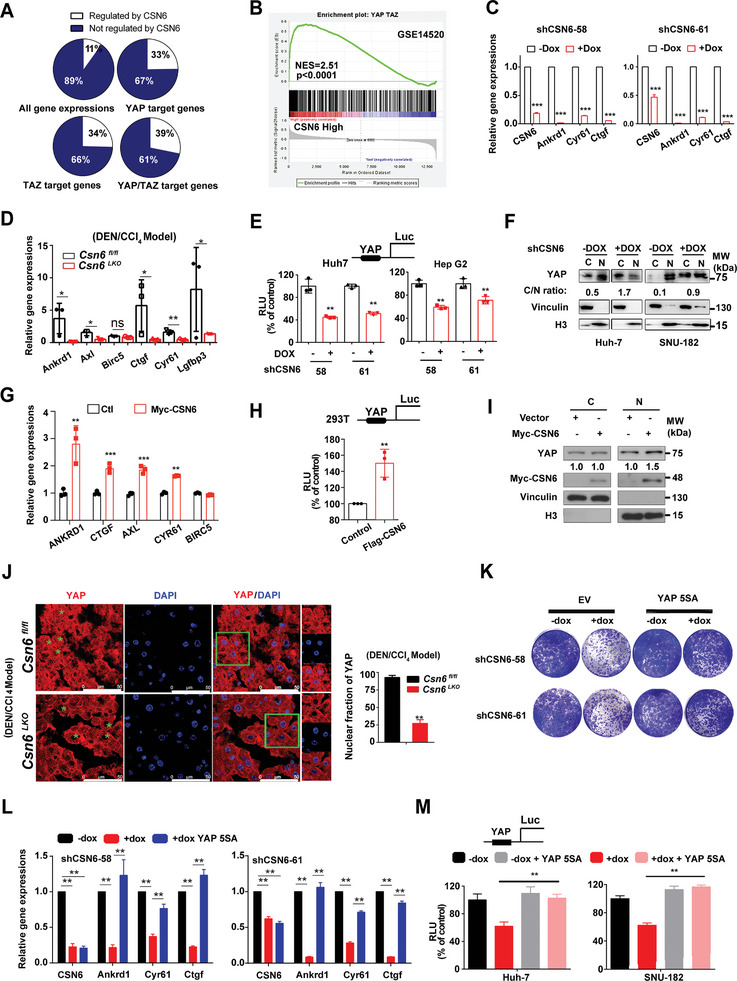
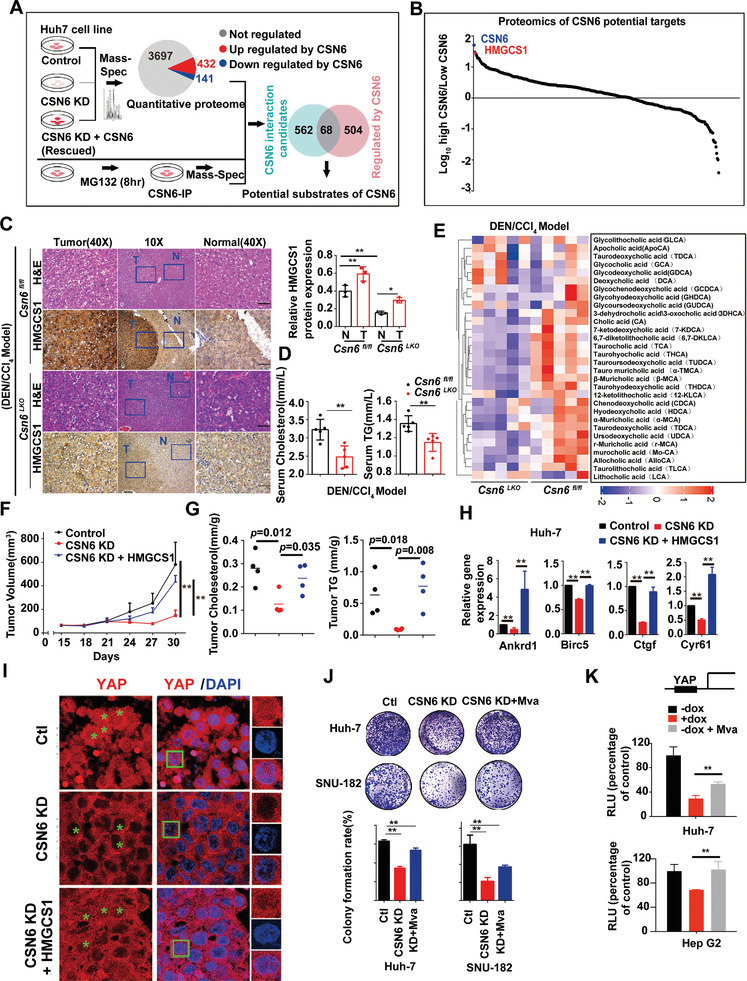
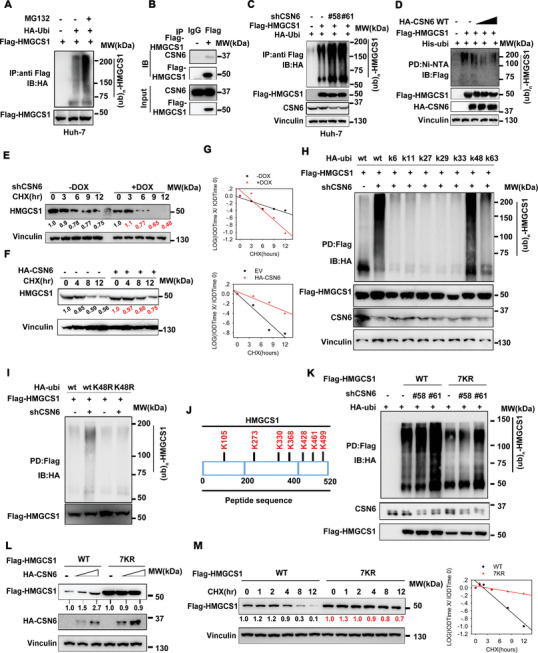
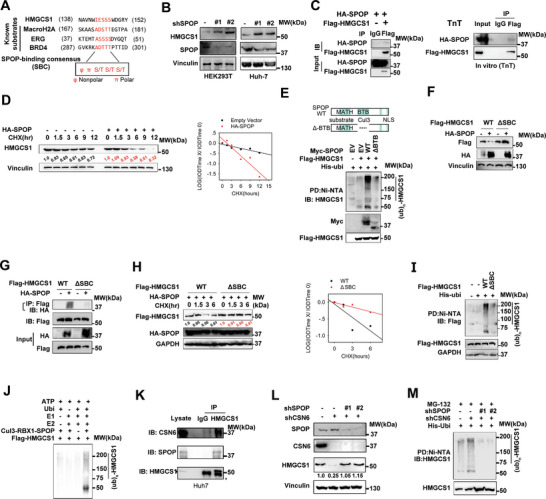
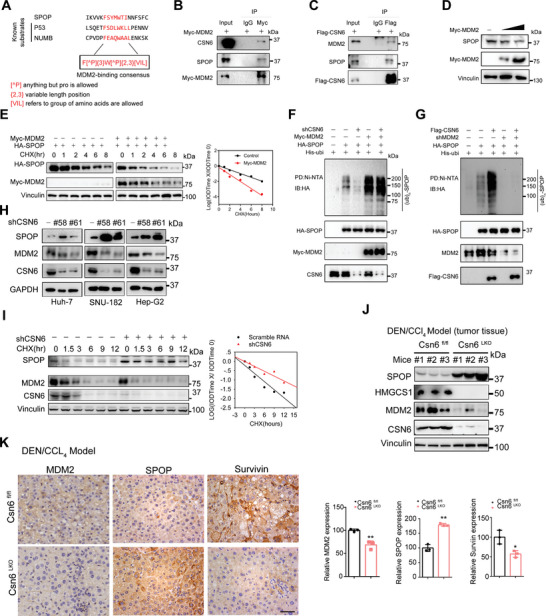
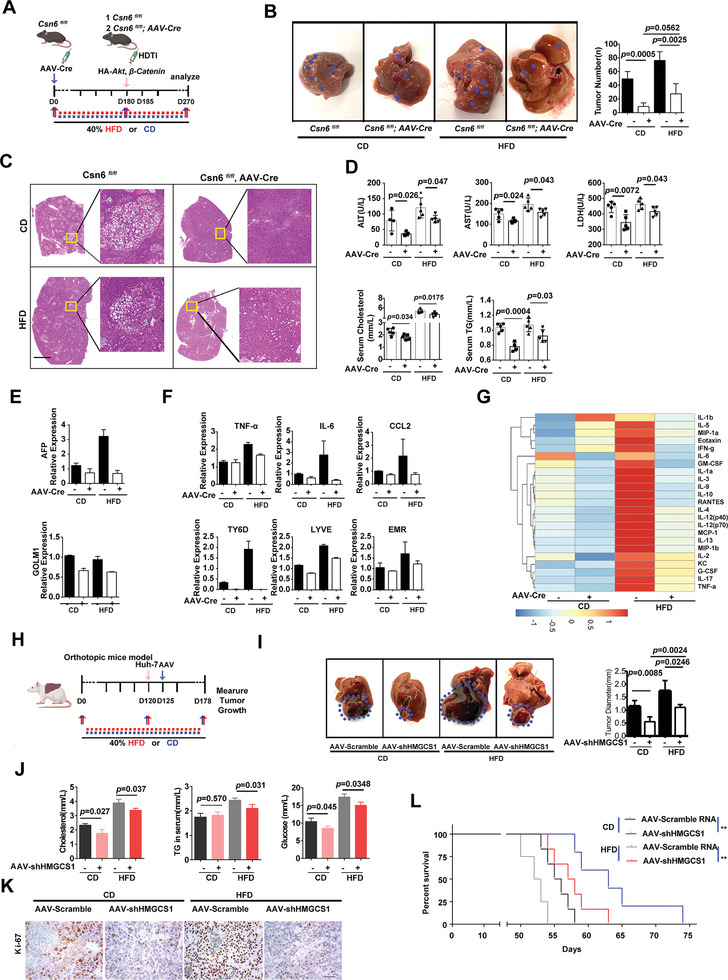
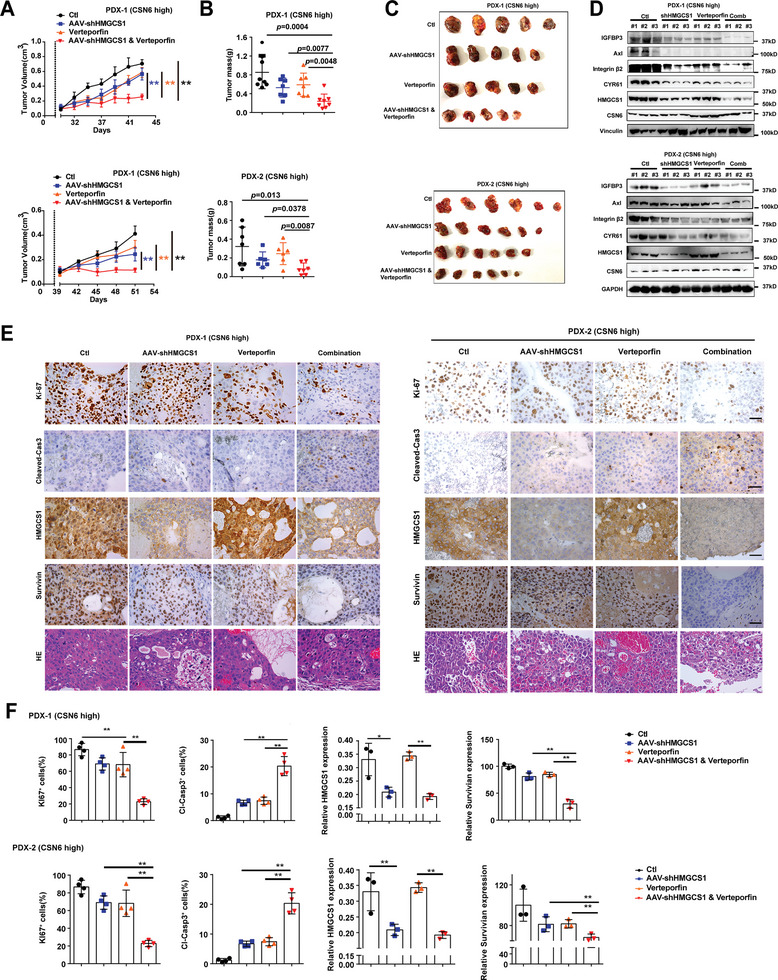
Cholesterol metabolism has important roles in maintaining membrane integrity and countering the development of diseases such as obesity and cancers. Cancer cells sustain cholesterol biogenesis for their proliferation and microenvironment reprograming even when sterols are abundant. However, efficacy of targeting cholesterol metabolism for cancer treatment is always compromised. Here it is shown that CSN6 is elevated in HCC and is a positive regulator of hydroxymethylglutaryl-CoA synthase 1 (HMGCS1) of mevalonate (MVA) pathway to promote tumorigenesis. Mechanistically, CSN6 antagonizes speckle-type POZ protein (SPOP) ubiquitin ligase to stabilize HMGCS1, which in turn activates YAP1 to promote tumor growth. In orthotopic liver cancer models, targeting CSN6 and HMGCS1 hinders tumor growth in both normal and high fat diet. Significantly, HMGCS1 depletion improves YAP inhibitor efficacy in patient derived xenograft models. The results identify a CSN6-HMGCS1-YAP1 axis mediating tumor outgrowth in HCC and propose a therapeutic strategy of targeting non-alcoholic fatty liver diseases- associated HCC.
Keywords: HFD; YAP1; cholesterol metabolism; hepatocellular carcinoma(HCC).
© 2024 The Authors. Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- a) Luo J., Yang H., Song B. L., Nat. Rev. Mol. Cell Biol. 2020, 21, 225; - PubMed
- b) Cao Z., Fan‐Minogue H., Bellovin D. I., Yevtodiyenko A., Arzeno J., Yang Q., Gambhir S. S., Felsher D. W., Cancer Res. 2011, 71, 2286; - PMC - PubMed
- c) Lei K., Kurum A., Kaynak M., Bonati L., Han Y., Cencen V., Gao M., Xie Y. Q., Guo Y., Hannebelle M. T. M., Wu Y., Zhou G., Guo M., Fantner G. E., Sakar M. S., Tang L., Nat. Biomed. Eng. 2021, 5, 1411; - PMC - PubMed
- d) Liao P., Wang W., Wang W., Kryczek I., Li X., Bian Y., Sell A., Wei S., Grove S., Johnson J. K., Kennedy P. D., Gijon M., Shah Y. M., Zou W., Cancer Cell 2022, 40, 365. - PMC - PubMed
MeSH terms
Substances
Grants and funding
- 2020YFA0803300/National Key Research and Development Program
- 2018YFC0910300/National Key Research and Development Program
- 82203243/National Natural Science Foundation of China
- 82204411/National Natural Science Foundation of China
- 81971999/National Natural Science Foundation of China
- 2023A1515030245/Natural Science Foundation of Guangdong Province
- 2020A1515110076/Natural Science Foundation of Guangdong Province
- "President Specific Funding" of The Sixth Affiliated Hospital, Sun Yat-sen University
- 2020B1111170004/Guangdong Provincial Clinical Research Center for Digestive Diseases
- National Key Clinical Discipline
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials