

Micropillar arrays, wide window acquisition and AI-based data analysis improve comprehensiveness in multiple proteomic applications
- PMID: 38310095
- PMCID: PMC10838342
- DOI: 10.1038/s41467-024-45391-z
Micropillar arrays, wide window acquisition and AI-based data analysis improve comprehensiveness in multiple proteomic applications
Abstract
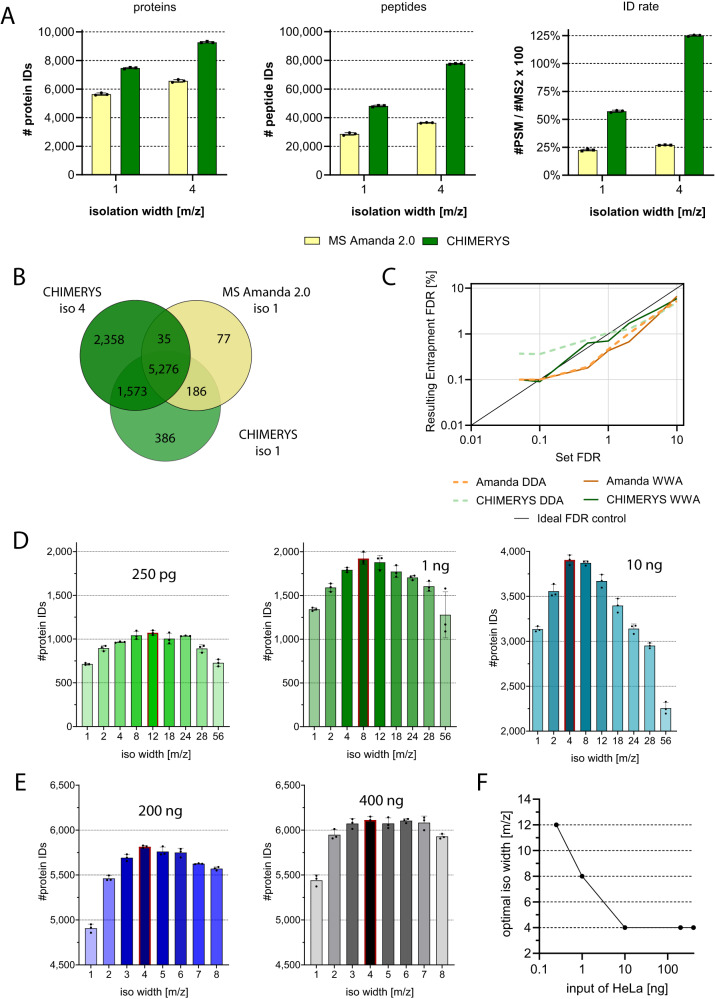
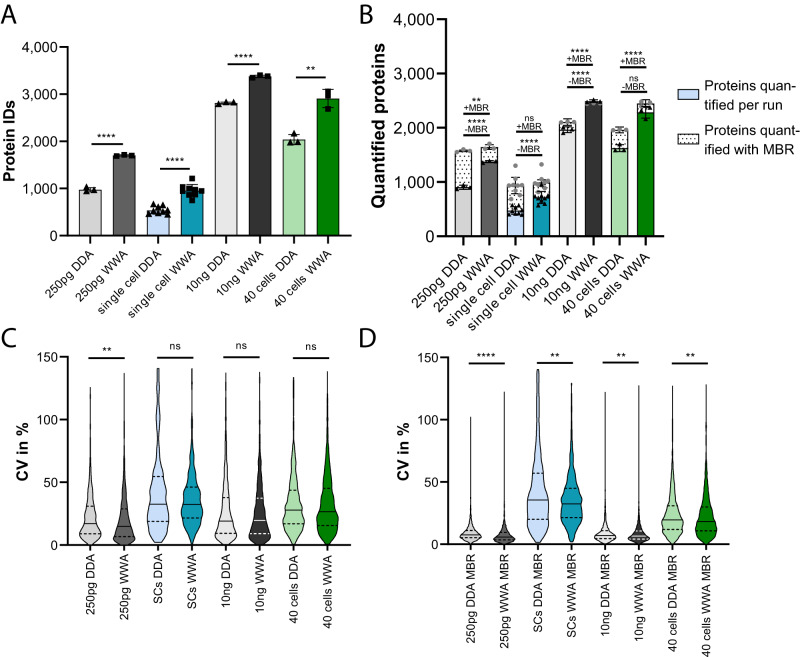
Comprehensive proteomic analysis is essential to elucidate molecular pathways and protein functions. Despite tremendous progress in proteomics, current studies still suffer from limited proteomic coverage and dynamic range. Here, we utilize micropillar array columns (µPACs) together with wide-window acquisition and the AI-based CHIMERYS search engine to achieve excellent proteomic comprehensiveness for bulk proteomics, affinity purification mass spectrometry and single cell proteomics. Our data show that µPACs identify ≤50% more peptides and ≤24% more proteins, while offering improved throughput, which is critical for large (clinical) proteomics studies. Combining wide precursor isolation widths of m/z 4-12 with the CHIMERYS search engine identified +51-74% and +59-150% more proteins and peptides, respectively, for single cell, co-immunoprecipitation, and multi-species samples over a conventional workflow at well-controlled false discovery rates. The workflow further offers excellent precision, with CVs <7% for low input bulk samples, and accuracy, with deviations <10% from expected fold changes for regular abundance two-proteome mixes. Compared to a conventional workflow, our entire optimized platform discovered 92% more potential interactors in a protein-protein interaction study on the chromatin remodeler Smarca5/Snf2h. These include previously described Smarca5 binding partners and undescribed ones including Arid1a, another chromatin remodeler with key roles in neurodevelopmental and malignant disorders.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
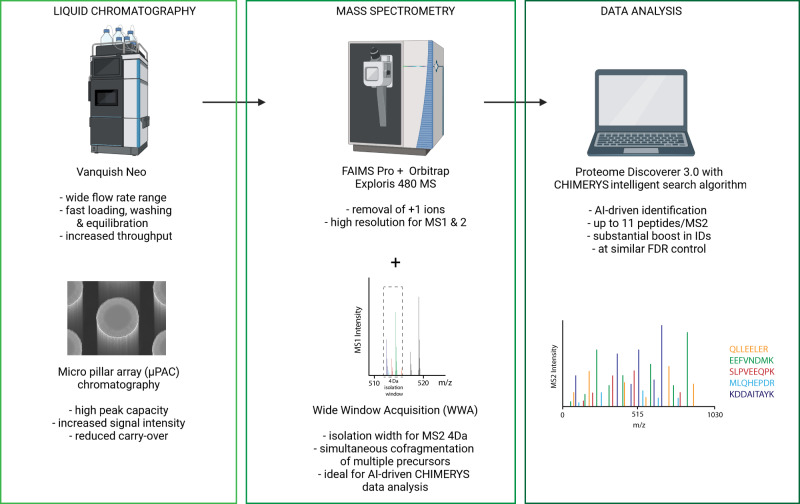
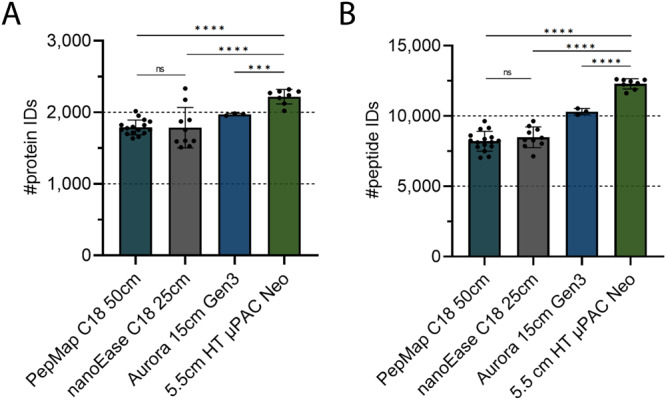
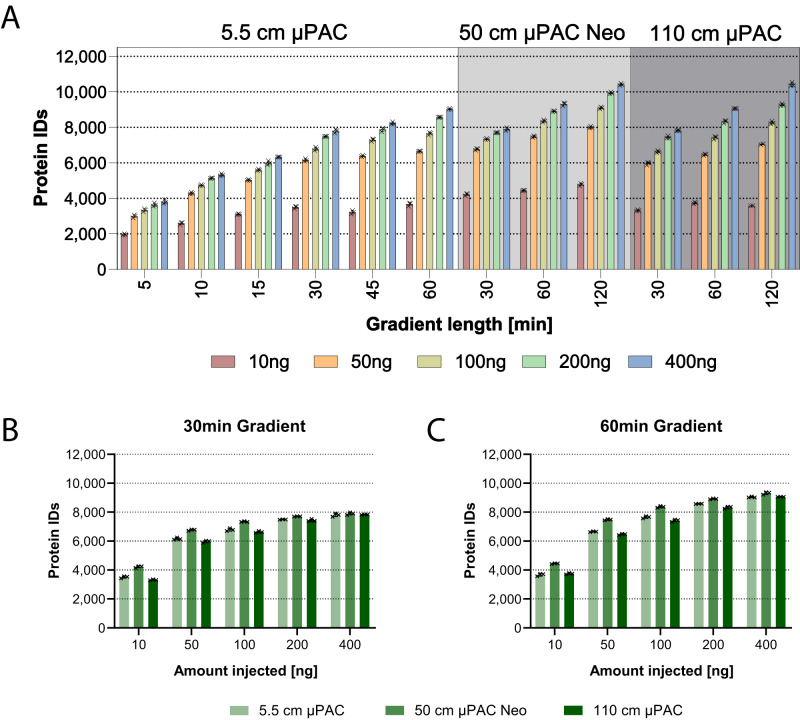
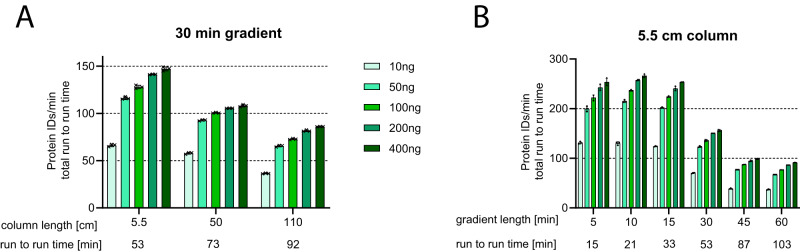

References
MeSH terms
Substances
Grants and funding
- P35045-B/Austrian Science Fund (Fonds zur F&#x00F6;rderung der Wissenschaftlichen Forschung)
- P35045-B/Austrian Science Fund (Fonds zur F&#x00F6;rderung der Wissenschaftlichen Forschung)
- P32054/Austrian Science Fund (Fonds zur F&#x00F6;rderung der Wissenschaftlichen Forschung)
- P33380/Austrian Science Fund (Fonds zur F&#x00F6;rderung der Wissenschaftlichen Forschung)
- LS20-079/Vienna Science and Technology Fund (Wiener Wissenschafts-, Forschungs- und Technologiefonds)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
